
Abstract
Thrombotic Microangiopathy (TMA) is a syndrome characterized by endothelial damage, leading to abnormal activation of coagulation, Microangiopathic Hemolytic Anemia (MAHA), thrombocytopenia, and organ damage. Within the spectrum of TMA, Complement-Mediated Thrombotic Microangiopathy (CM-TMA) is a subtype that can be classified as primary (idiopathic or inherited) or secondary to complement-activating conditions. In this case report, we present the case of a woman in her mid-50s with a medical history remarkable for Systemic Lupus Erythematosus (SLE) and Coexisting Cholangiocarcinoma (CCA). After initiating chemotherapy with gemcitabine/cisplatin, the patient developed MAHA, severe thrombocytopenia, and acute renal failure. Extensive evaluations ruled out thrombotic Thrombocytopenic Purpura (TTP) and Shiga toxin-mediated TMA. Despite initial treatment with corticosteroids and daily plasma exchange, the patient’s condition did not show significant improvement. This led to the suspicion of secondary CM-TMA, which prompted the initiation of Ravulizumab, a complement C5 inhibitor. The administration of Ravulizumab resulted in a remarkable and rapid normalization of renal function, hemolysis parameters, and platelet count. This case highlights the challenges in diagnosing and managing TMA and emphasizes the importance of suspecting secondary CM-TMA in patients with complement-activating conditions diagnosed with TMA, in which thrombotic thrombocytopenic purpura and Shiga toxin-mediated TMA are ruled out, and who lack a response to conventional treatment. It also demonstrates how early initiation of C5 inhibitor therapy is crucial to improving outcomes in these patients.
Keywords: Complement-mediated thrombotic microangiopathy; Complement inhibitor therapy; Thrombotic microangiopathy
Case Report
A woman in her mid-50s with a past medical history significant for well-controlled SLE and hypothyroidism presented initially with constipation and generalized abdominal pain. CT abdomen/pelvis showed a poorly defined left lobe liver lesion. Left lateral partial hepatectomy was performed. Pathology was consistent with CCA. Post-operatively, she was started on adjuvant chemotherapy with Cisplatin and Gemcitabine (days 1 and 8 every 21 days). The patient was noted to have thrombocytopenia (23 x 103/μL) and neutropenia (1.6 x 103/μL) after the first cycle, so the chemotherapy dose was reduced. Chemotherapy was later stopped after six cycles when the patient was noted to have persistent severe thrombocytopenia (12 x 103/μL), anemia with hemoglobin decreased to 6.8 g/dL, and severe fatigue. No arthralgias, skin changes, or oral ulcers were reported.
The patient was also found to have schistocytes on a peripheral smear (Figure 1), increased lactate dehydrogenase level, decreased haptoglobin, and acute kidney injury, with creatinine levels increased to 2.5 mg/dL. Further workup revealed a negative stool culture for Shiga Toxin-producing Escherichia Coli (STEC), normal pretreatment ADAMTS13 activity, increased rheumatoid factor, decreased complement component 3 and 4 (C3/C4) levels, and positive Sjogren's SS-A antibody. Shiga toxin-mediated TMA was ruled out after stool culture returned negative for Shiga Toxin-producing Escherichia Coli (STEC). TTP was also excluded based on normal pretreatment ADAMTS13 activity. The patient's thrombocytopenia, anemia, and renal failure continued to worsen despite discontinuing the offending chemotherapeutic agent, and no significant recovery was observed after starting corticosteroid treatment and undergoing four sessions of daily Plasma Exchange (PEX). Genetic mutation analysis for complement-regulatory protein abnormalities, which could have helped rule out primary CM-TMA, was not conducted in this patient. The diagnosis of secondary CM-TMA was made based on the patient's medical history of SLE and cholangiocarcinoma, recent treatment with Gemcitabine, and the lack of response to corticosteroids and PEX.
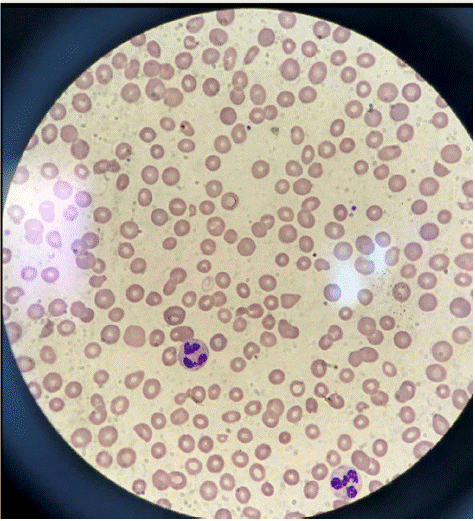
Figure 1: Peripheral blood film showing numerous schistocytes.
Ravulizumab, a monoclonal antibody targeting and inhibiting complement component 5, was initiated. Our patient’s response to this treatment was remarkable, resulting in the rapid normalization of platelet count, hemoglobin levels, and renal function. Since then, the patient has continued to receive Ravulizumab infusions every eight weeks, completing a total of 12 cycles up to this point, and has consistently shown a positive response to the treatment.
Discussion
Thrombotic Microangiopathy (TMA) is a pathological condition caused by the formation of microvascular thrombi, commonly presenting with thrombocytopenia and Microangiopathic Hemolytic Anemia (MAHA). Despite having diverse etiologies, these disorders share similar clinical presentations.
Thrombotic Thrombocytopenic Purpura (TTP) is a type of TMA characterized by the formation of widespread von Willebrand Factor (vWF)-platelet thrombi in arterioles and capillaries of multiple organs. This condition results from impaired function of the ADAMTS13 enzyme, responsible for cleaving vWF into fragments [1]. Shiga toxin-mediated TMA is caused by the action of a bacterial toxin, leading to endothelial damage and subsequent organ dysfunction. Drug-induced TMA is associated with certain medications such as mitomycin, gemcitabine, tacrolimus, and sirolimus.
Another distinct form of TMA is complement-mediated Thrombotic Microangiopathy (CM-TMA), formerly known as atypical hemolytic uremic syndrome (aHUS). CM-TMA is driven by dysregulation of the complement system, which leads to complement deposition on endothelial cells. As a consequence, endothelial cell dysfunction occurs, leading to the formation of microvascular thrombi and the creation of schistocytes through red blood cell shearing, resulting in non-immune hemolytic anemia, thrombocytopenia and renal impairment.
CM-TMA can be categorized into two main types: primary (idiopathic or inherited) and secondary to complement-activating conditions. Primary CM-TMA results from mutations in genes that encode complement regulatory proteins or the presence of Complement Factor H (CFH) autoantibodies. These events result in the triad of non-immune hemolytic anemia, renal impairment, and thrombocytopenia. The analysis of mutations in complement-related genes or the measurement of antibodies to complement factors can be helpful in diagnosing primary CM-TMA [2,3]. Secondary CM-TMA is triggered by complement-activating conditions, including sepsis, hematopoietic stem cell or solid organ transplantation, autoimmune disorders (particularly Systemic Lupus Erythematosus (SLE) and Systemic Sclerosis), malignant hypertension, viral infections (i.e. HIV, H1N1 influenza virus), and malignancies [3,4].
The diagnosis and management of TMA can be challenging, especially when multiple potential underlying etiologies are present. One limitation of this case is that genetic testing to identify mutations in complement regulatory proteins and autoantibodies against complement factors, which could have ruled out primary CM-TMA, was not conducted. However, secondary CM-TMA was suspected after ruling out TTP and Shiga toxin-mediated TMA, as the patient did not respond to conventional therapies, and also considering her pertinent medical history. Firstly, the patient had a history of SLE, an autoimmune disorder known to be associated with an elevated risk of TMA, as previously described. Secondly, the patient was diagnosed with CCA, and malignant conditions have also been linked to TMA [5,6]. Additionally, at the time of TMA diagnosis, the patient had recently started chemotherapy with Gemcitabine, which has been reported to cause drug-induced TMA [7].
Systemic Lupus Erythematosus (SLE), can lead to endothelial cell damage and complement dysregulation, both of which are important components in the pathogenesis of TMA. Malignancies can also lead to TMA secondary to the release of procoagulant factors and tumor-related endothelial cell damage. Furthermore, specific chemotherapeutic agents, such as Gemcitabine, have been reported to induce drug-induced TMA. This adverse effect can occur either as an immune-mediated reaction or as a result of toxic reactions related to the dose or duration of treatment.
Our patient's positive response to subsequent treatment with Ravulizumab, a complement C5 inhibitor, further provided support for the potential involvement of the complement pathway in the pathogenesis of TMA in this case. Eculizumab and Ravulizumab are monoclonal antibodies that inhibit complement component 5 (C5), approved for the treatment of CM-TMA [8]. Several cases of TMA associated with gemcitabine and lupus nephritis successfully treated with eculizumab have been reported [9,10].
Treatment with Eculizumab consists of an intravenous infusion administered every 2 weeks. Ravulizumab is a new complement C5 inhibitor that produces immediate, complete, and sustained inhibition of C5, with an 8-week dosing interval [11]. Before the development of C5 inhibitor therapy, up to two-thirds of patients with CM-TMA died or progressed to end-stage renal disease in the first year after their initial presentation.
The use of C5 inhibitors, such as Ravulizumab or Eculizumab, in cases of SLE-associated TMA has been documented in the medical literature. Previous reports have demonstrated successful outcomes with C5 inhibitor therapy in these patients, further supporting the potential involvement of the complement pathway in the pathogenesis of this condition [12].
Lifelong C5 inhibitor therapy, specifically Eculizumab, was initially recommended for all patients with CM-TMA; however, recent evidence suggests that discontinuation of treatment could be considered after 6 to 12 months of treatment, with at least 3 months of normalization of renal function or stabilization of chronic renal disease. A period beyond 12 months should be considered if a complement mutation is detected or anti-CFH autoantibodies are present, or if the patient has a history of a prior CM-TMA episode or renal transplant. For patients with recognized complement-activating conditions, treatment should be continued until the condition has resolved [13].
Conclusion
This case report emphasizes the critical role of prompt diagnosis and timely intervention in patients with suspected secondary CM-TMA. For patients presenting with TMA, after excluding TTP and Shiga toxin-mediated TMA and encountering a poor response to corticosteroids/plasma exchange, and with suspicion of primary or secondary CM-TMA, it is important to consider treatment with C5 inhibitor therapy.
Early initiation of C5 inhibitor therapy, such as Ravulizumab or Eculizumab, in patients with suspected secondary CM-TMA, has shown to be remarkably effective in improving outcomes in these patients, leading to rapid normalization of platelet count, hemoglobin levels, and renal function.
References
- Kappler S, Ronan-Bentle S, Graham A. Thrombotic Microangiopathies (TTP, HUS, HELLP). Hematol Oncol Clin North Am. 2017; 31: 1081-1103.
- Raina R, Krishnappa V, Blaha T, Kann T, Hein W, Burke L, et al. Atypical Hemolytic-Uremic Syndrome: An Update on Pathophysiology, Diagnosis, and Treatment. Ther Apher Dial. 2019; 23: 4-21.
- Palma LMP, Sridharan M, Sethi S. Complement in Secondary Thrombotic Microangiopathy. Kidney Int Rep. 2021; 6: 11-23.
- Laurence J, Haller H, Mannucci PM, Nangaku M, Praga M, Rodriguez de Cordoba S. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol. 2016; 14: 2-15.
- López Rubio ME, Rodado Martínez R, Illescas ML, Mateo Bosch E, Martinez Díaz M, de la Vara Inesta L, et al. Gemcitabine-induced hemolytic-uremic syndrome treated with eculizumab or plasmapheresis: two case reports. Clin Nephrol. 2017; 87: 100-106.
- Figueiredo CR, Escoli R, Santos P, Sofia F, Lopes K. Thrombotic microangiopathy in a patient with systemic lupus erythematosus and anti-factor H autoantibodies. CEN Case Rep. 2022 Feb;11(1):26-30.
- Hasan A, Jain AG, Naim H, Munaf A, Everett G. Drug-induced Thrombotic Microangiopathy Caused by Gemcitabine. Cureus. 2018; 10: e3088.
- Syed YY. Ravulizumab: A Review in Atypical Haemolytic Uraemic Syndrome. Drugs. 2021; 81: 587-594.
- Grall M, Provôt F, Coindre JP, Pouteil-Noble C, Guerrot D, Benhamou Y, et al. Efficacy of eculizumab in gemcitabine-induced thrombotic microangiopathy: experience of the French Thrombotic Microangiopathies Reference Centre. Blood 2016; 128: 136.
- Park MH, Caselman N, Ulmer S, Weitz IC. Complement-mediated thrombotic microangiopathy associated with lupus nephritis. Blood Adv. 2018; 2: 2090-2094.
- Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019; 133: 540-549.
- Yamaguchi M, Mizuno M, Kitamura F, Iwagaitsu S, Nobata H, Kinashi H, et al. Case report: Thrombotic microangiopathy concomitant with macrophage activation syndrome in systemic lupus erythematosus refractory to conventional treatment successfully treated with eculizumab. Front Med (Lausanne). 2023; 9: 1097528.
- Laurence J. Defining treatment duration in atypical hemolytic uremic syndrome in adults: a clinical and pathological approach. Clin Adv Hematol Oncol. 2020; 18: 221-230.