
Abstract
Background: MBT1805 is a novel pan-Peroxisome Proliferator-Activated Receptor (PPAR) agonist.
Materials and Methods: In vitro, transfection and luciferase assays tested EC50 values of MBT1805. In vivo, hypoglycemic and hypolipidemic effects of MBT1805 were observed in db/db mice compared with Rosiglitazone.
Results: In vitro, MBT1805 activates human PPARα, PPARγ and PPARδ with EC50 values of 8.46μM, 11.94μM, 11.15μM, respectively. Results showed that the bodyweight of db/db mice treated with MBT1805 was not changed. By contrast, Rosiglitazone-treated mice showed significant weight gain (p<0.05). MTB1805 decreased blood glucose level without causing noticeable hepatocytes damage.
Conclusion: The novel balanced pan-PPAR agonist, MBT1805 has moderate hypoglycemic and hypolipidemic effects, and does not cause weight gain, hepatocyte damage and hepatic lipid deposition. These experimental results indicate that MBT1805 is safe in the treatment of type 2 diabetes.
Keywords: Peroxisome proliferator-activated receptor; PPARs agonist; Diabetes; MBT1805; Rosiglitazone
Abbreviation
PPARs: Peroxisome Proliferator-Activated Receptors; T2DM: Type 2 Diabetes Mellitus; OGTT: Oral Glucose Tolerance Test; AUC: Area Under the Curve; TC: Total Cholesterol; TG: Triglyceride; Tbil: Total Bilirubin; FFAS: Free Fatty Acids; EC50: Half Maximal Effective Concentration
Introduction
Rosiglitazone is FDA approved for the treatment of Type 2 Diabetes Mellitus (T2DM). It effectively lowers glucose levels by improving targets cells response to insulin without enhancing insulin release by pancreatic beta cells [1]. However, side effects, including weight gain, fluid retention, and heart failure constrain the use of TZDs. Troglitazone has been withdrawn from the market due to liver toxicity [2]. Peroxisome Proliferator-Activated Receptors (PPARs) have exhibited potential benefits against diabetes. Balanced regulation of PPARα, PPARγ and PPARδ may be beneficial, while minimizing harmful side effects.
PPARs are ligand dependent transcription factors, belonging to the nuclear hormone receptor superfamily. Three PPAR isoforms are known, PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3), which are all involved in fat and carbohydrate metabolism and homeostasis. PPARs also influence proliferation and differentiation, inflammation, vascular biology and cancer [3]. The 3 PPAR isoforms have distinct but complementary physiological functions due to different tissue distribution, ligand sensitivities and target genes [4]. PPARα is highly expressed in tissues rich in Fatty Acid Oxidation (FAO), including liver, heart, skeletal muscle, brown adipose tissue, and kidney. PPARα activation stimulates fatty acid and triglyceride metabolism. PPARβ/δ is broadly expressed and is crucial for the activation of fatty acids in skeletal muscles. Its expression is markedly enhanced by fasting and exercise [5]. PPARβ/δ activation also improves hepatic insulin response by suppressing hepatic gluconeogenesis at the postprandial stage. There are 2 PPARγ isoforms, PPARγ1 and PPARγ2. PPARγ activation elevates insulin sensitivity in the whole body [4,6-9]. A variety of endogenous ligands activate PPARs and the degree of receptor activation depends on the balance between ligands production and inactivation. Endogenous ligands may come from diet, de novo Lipogenesis (DNL), or lipolysis, and they include n-3 and n-6 Fatty Acids (FAs), eicosanoids, some endocannabinoids and phospholipids [6-8]. The synthetic ligands also potently modulate PPARs function. Except for PPARβ/δ, for which selective modulators have not entered the clinic, selective PPARα and PPARγ agonists are used clinically. Moreover, dual, and pan-PPAR agonists are under development and will enable exploration of PPAR complementary roles. Treatment of dyslipidemia and type 2 diabetes is relatively advanced. Fibrates (Fenofibrate and Bezafibrate) are often used in combination with satins to treat atherogenic hyperlipidemia and hypertriglyceridemia [10,11]. Similarly, TZDs are used to treat T2DM [12]. Ideally, PPAR modulators should possess superior bioavailability and pharmacokinetics, with minimal off-target and side effects [13].
Multiple novel drugs targeting PPARs are under development. Here, we treated db/db mice with MBT1805, a novel pan-PPARs agonist and evaluated its hypoglycemic and hypolipidemic effect, as well as side effects, relative to rosiglitazone. The db/db mice carry spontaneous mutations in the gene encoding the long isoform of leptin receptor in hypothalamus, resulting in persistent and severe glucose intolerance, hyperglycemia and hyperinsulinemia. This mouse is an established model of T2DM [14,15].
Materials and Methods
Reagents
Hepatoma cell line was purchased from ATCC. FuGENE6 transfection reagent was purchased from Roche (Cat No11814443001). MBT1805 (Cat No190509), fenofibrate and rosiglitazone maleate (HBW190503-6) were supplied by Beijing JK HuaYuan Med Tech Company LTD free for charge (Beijing, China). Tween80 (Cat NoA0341586) was purchased from Acros Organics. CMC-Na (Cat No16774) was purchased from jingchun (Shanghai, China). DMSO (Cat NoD8371) were purchased from Solarbio (Beijing, China). Sterile water (Cat No180416205) was purchased from Aixide (Guangdong, China). Anhydrous glucose (Cat No20140106) was purchased from Guoyao (Shanghai, China).
In vitro transfection and luciferase assays
Our method was adopted from Zhibin Li [16]. A Nuclear Receptor DNA incorporating sequence (NRE) was cloned into pcDNA3.1 expression vector, upstream of luciferase gene, and insertion confirmed by sequencing (data not shown). When PPARs are activated, the binding with NRE is enhanced, thus increasing expression of Luciferase gene in the downstream. Luciferase intensity indirectly reflect the activation of PPARs. HepG2 cells were cultured in DMEM supplemented with 10% FBS, at 37ºC, in a humidified incubator with 5% CO2. Cells were seeded in 96-well plates the day before transfection to allow for 50-80 % confluence at transfection. They were then transfected with indicated plasmids using FuGENE6 according to manufacturer instructions. A GFP plasmid was transfected as negative control. 24 hours after transfection, media was replaced with fresh complete media. MBT1805 and fenofibrate were dissolved in DMSO. Cells were treated with MBT1805 and fenofibrate at various final concentrations. As negative control, vehicle only (DMSO) was used at a final concentration of 0.1%. Cells were cultured for 24 hours before they were directly harvested into cell lysis buffer. Luciferase and GFP activities were measured, and values from all test wells normalized to GFP. Results were presented as fold changes relative to the negative control. The value obtained was directly proportional to activation strength.
Animal treatment
34 db/db and 12 C57BL/6J mice (male), 9-10-weeks old, were purchased from Jicuiyaolkang (Jiangsu, China). 8 healthy C57 mice were used as blank control group. The 34 db/db mice were randomly divided into 4 groups, model control (vehicle), rosiglitazone (10mg/ kg), and MBT1805 (15mg/kg and 30mg/kg), 8-9 mice per group. Mice were maintained under controlled temperature and humidity (23±2ºC, 40-70 % respectively) and a 12h light/dark cycle. The animals had access to normal chow and water ad libitum. Where indicated, mice were fasted according to our animal protocol. All mice were raised adaptively for 13 days. After the adaption period, blood glucose levels in experimental animals were taken 4h after fasting. Compounds were dissolved at specified concentrations in a pre-formulated solvent of 0.05% Tween80/0.5% CMC-Na in sterile H2O. Drugs were administered once daily for 14 days. Mouse weight was taken twice weekly. On the 14th day, mice were fasted for 4h, and then treated. 1h later, blood samples were collected by tail bleeding and blood glucose measured.
Oral glucose tolerance test
On the 15th day, 16h after fasting, animals were weighted and their blood glucose measured using a blood glucose meter (Xpress) at 0min. Glucose solution (2mg/kg) was immediately administered by oral gavage at the time of compounds administration. Blood glucose was measured 30, 60, 120 and 180 min later, and the area under blood glucose time curve (AUC) calculated.
Serum and organ collection
Animals were anesthetized with CO2 and blood collected from the heart into 1.5mL centrifuge tubes and stored for 30min at room temperature. They were then centrifuged at 12000rpm for 5min. The serum was then collected, and serum TC, TG, ALT, AST, Tbil, FFA and CREA detected using an automatic biochemical analyzer (Rili, 76000). The animals were then euthanized by inhaling excessive CO2. Livers were then collected, weighted and liver coefficient calculated. About 150mg of liver was snap-frozen in liquid and stored at -80ºC for TC and TG detection.
Statistical analysis
Statistical analysis was done using SPSS version 21 (Chicago, IL, USA) and data plotted on GraphPad 8.0 (GraphPad Software, La Jolla, CA). Quantitative date is presented as mean ± SEM. Unpaired t test was used for comparisons between 2 groups. P value ≤0.05 indicates statistical significance.
Results
Separated EC50 values of MBT1805 or fenofibrate in activating PPARs
Half maximal Effective Concentration (EC50) is an important index for accessing the pharmacological activity of compounds. PPARs activation using 6 concentrations of MBT1805 or fenofibrate was assessed. We iteratively calculated and fitted the concentration effect curve, and calculated corresponding EC50. We observe that MBT1805 markedly activates PPARα, PPARγ and PPARδ. EC50 values for PPARα, PPARγ and PPARδ are 8.455μM, 11.94μM and 11.15μM, respectively (Table 1). It was worth noting that lower luciferase values were detected when PPARγ and PPARδ was activated by higher MBT1805 dosage (100μM). This may be because activation of PPARs by MBT1805 triggers intracellular negative feedback regulation. Fenofibrate, a selective PPARα agonist, exhibited a significantly lower EC50 value of PPARα relative to the other 2 receptors, (Table 2). MBT1805 activated PPARα more potently than fenofibrate at the same concentration. The ability of MBT1805 to activate PPARγ and PPARδ was significantly higher than that of fenofibrate.
![]()
EC50 (μM)
0.3μM
1μM
3μM
10μM
30μM
100μM
PPARα
1.36 ± 0.05
1.83 ± 0.13
2.18 ± 0.18
2.36 ± 0.16
2.87 ± 0.32
3.58 ± 0.28
8.455
PPARγ
0.98 ± 0.08
1.09 ± 0.20
1.33 ± 0.06
1.71 ± 0.14
4.50 ± 0.65
2.85 ± 0.26
11.94
PPARδ
0.91 ± 0.13
1.02 ± 0.09
1.24 ± 0.14
1.51 ± 0.55
3.94 ± 0.55
2.89 ± 0.18
11.15
Table 1: Separated EC50 values of MBT1805 activating PPARs.
![]()
EC50 (μM)
1μM
3μM
10μM
30μM
100μM
300μM
PPARα
1.15 ± 0.06
1.45 ± 0.11
1.89 ± 0.12
2.44 ± 0.08
2.66 ± 0.11
2.73 ± 0.18
8.511
PPARγ
1.07 ± 0.08
1.18 ± 0.12
1.37 ± 0.18
1.76 ± 0.17
5.17 ± 0.67
8.35 ± 0.51
104.7
PPARδ
0.95 ± 0.16
1.04 ± 0.12
1.07 ± 0.07
1.20 ± 0.08
1.71 ± 0.07
2.46 ± 0.13
125.9
Table 2: Separated EC50 values of Fenofibrate activating PPARs.
Clinical observations and liver coefficient calculation
All experimental mice survived, and seemed normal during the experiment. Figure 1a shows bodyweight changes for animals in various groups during the experiment. Mice treated with rosiglitazone exhibited significant weight gain by day 14 and 15 (p<0.05). Relative to vehicle treated animals, those treated with MBT1805 did not show significant weight change (p>0.05). Interestingly, liver coefficient (Table 3) for all treated mice were higher relative to the vehicle treated group (p<0.001).
![]()
Liver Weight (g)
Liver coefficient (%)
Blank control
23.35 ± 0.30***
0.89 ± 0.01***
3.80 ± 0.05***
Vehicle
47.25 ± 1.93
2.44 ± 0.14
5.18 ± 0.25
Rosiglitazone 10mg/Kg
54.21 ± 1.39*
4.31 ± 0.09*
7.96 ± 0.17***
MBT1805 15mg/Kg
49.60 ± 0.88
4.02 ± 0.14
8.10 ± 0.23***
MBT1805 30mg/Kg
47.32 ± 1.69
4.05 ± 0.22
8.53 ± 0.29***
*p<0.05; ***p<0.001 vs. Vehicle.
Table 3: Liver coefficient calculation.
![]()
TG (mmol/gprog)
Blank control
0.01 ± 0.00**
0.12 ± 0.03***
Vehicle
0.02 ± 0.00
1.21 ± 0.20
Rosiglitazone 10mg/kg
0.15 ± 0.04*
MBT1805 15mg/kg
0.02 ± 0.01
0.92 ± 0.17
MBT1805 30mg/kg
0.02 ± 0.01
0.61 ± 0.14*
*p<0.05; ***p<0.001 vs. Vehicle.
Table 4: Hepatic TG and TC of various groups.
MBT1805 effectively reduces blood glucose
Blood glucose levels in the blank control group were significantly lower relative to the vehicle group on day 0 and day 14 (p<0.001). Compared with vehicle group, all treated db/db mice had lower blood glucose (p<0.05-0.01). Figure 1b shows changes in blood glucose level on day 0 and day 14. Fig1c shows OGTT results. Blood glucose levels and AUC values for the vehicle group during 0-180 min were significantly higher relative to the blank control group (p<0.001). Those of the rosiglitazone group were significantly reduced (p<0.01- 0.001). MBT1805 induced marked hypoglycemic effect after 30- 180min (p<0.05-0.01). There was no significant difference between the 15mg/kg group and the 30mg/kg group, except at 30min. Relative to rosiglitazone, MBT1805 has milder hypoglycemic effect.
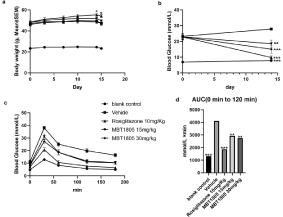
Figure 2: Bodyweight and Blood glucose changes of various groups. a)
Bodyweight values of rosiglitazone group significantly higher than that of
vehicle group (p<0.05) in day 14 and day 15. Bodyweight values of blank
control group of all timepoints are significantly lower compared with vehicle
group (p<0.001, not shown). b) Fasting blood glucose values of treated db/db
mice reduced compared with vehicle group (p<0.01-0.001). c,d) OGTT result
and AUC values. MBT1805 reduced blood glucose mildly, *p<0.05, **p<0.01,
***p<0.001 vs. Vehicle.
15mg/kg MBT1805 exhibited best efficacy in lowering Tbil, without hepatocyte damage
Higher lipid and liver injury occurred in db/db mice of the vehicle group, and serum TC, TG, ALT and Tbil were significantly elevated (p<0.05-0.001) relative to the blank control group. However, serum CREA levels were significantly lower (p<0.001), which is attributable to reduced activity by obese mice. At 30mg/kg, MBT1805 significantly reduced serum TC and TG levels (p<0.01, Figure 2c), and the lowest Tbil levels were induced by 15mg/kg MBT1805. Rosiglitazone also reduced serum TC, TG and Tbil, but elevated ALT and AST (p<0.01-0.001), suggesting hepatocyte damage. Lower FFAS levels were observed with higher MBT1805 dose, while rosiglitazone greatly reduced FFAS to levels lower than the blank control group. CREA levels in treated mice were higher than in the vehicle treated group, but lower relative to blank control group.
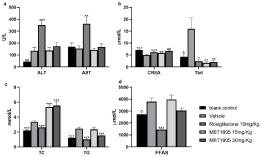
Figure 2: Serum biochemistry. a) ALT and AST significantly increase in
rosiglitazone group. b) CREA values of treated groups are higher than vehicle
group (p<0.01-0.001), Tbil value of MBT1805 at 15mg/Kg is the lowest. c)
MBT1805 reduces TG but elevates TC at 30mg/Kg. d) FFAS value reduces
with no significant difference; *p<0.05, **p<0.01, ***p<0.001 vs. Vehicle.
MBT1805 reduces hepatic TG, and not TC of db/db mice
Relative to blank control group mice, db/db mice treated with vehicle only exhibited hepatic lipid deposition (p<0.01-0.001). Relative to the vehicle, rosiglitazone significantly elevated hepatic TC and TG in db/db (p<0.05). TC values in MBT1805 groups were not reduced, but TG values were lower than in the vehicle group, especially in the 30mg/kg group (p<0.05). Suggesting that rosiglitazone increases lipid uptake in the liver and other tissues, lowering circulating TC, TG and FFAS. Increased lipid in hepatocytes may partly explain the ALT and AST elevation.
Discussion
Rosiglitazone, a selective PPARγ agonist, was used as positive control. We find that MBT1805 has mildly hypoglycemic effects without bodyweight gain or hepatocyte damage.
Rosiglitazone most effectively lowered fasting blood glucose and significantly improved OGTT results. PPARγ activation in adipocytes ensures optimal secretion of adipocytokines, containing adiponectin and leptin, which mediate insulin activity in peripheral tissues. These events maintain insulin sensitivity throughout the body [17]. We observed weight gain in rosiglitazone treated db/db mice (Figure 1a), which was consistent with previous findings [18,19]. However, MBT1805 treated db/db mice lacked obvious weight gain. The varying effects of MBT1805 and rosiglitazone on body weight suggest differences in fatty acids utilization. In addition, rosiglitazone treated mice showed varying degrees of liver damage, with higher ALT and AST levels relative to vehicle treated controls (Figure 2a). MBT1805 treated db/db mice had no obvious liver damage. Previous study suggested that full PPARγ agonism may be the cause of the side effects associated with glucose reduction [20]. PPARα activation enhances fatty acids oxidation, reducing fatty acids uptake, synthesis and associated high lipid mobilization in adipose tissues. PPARδ activation enhances fatty-acid-burning capacity in skeletal muscle, which is accompanied by a redistribution of fatty acid flux adipose tissue to skeletal muscles. The sum of PPARα and PPARδ activation offsets fat accumulation and weight gain induced by PPARγ agonism.
MBT1805 significantly lowered fasting blood glucose levels (p<0.01, Figure 2b). In the oral tolerance test, MBT1805 mildly lowered blood glucose in 30min, which was sharply reduced in rosiglitazone group. Rosiglitazone treatment for diabetes- induced hypoglycemic has been previously reported. Hypoglycemia was more frequently observed in women treated with TZDs relative to men [21]. A Meta-analysis revealed that adding rosiglitazone to insulin may help lower glucose levels in type 2 diabetes patients whose glucose levels are poorly controlled by insulin. However, this benefit is at the cost of higher total cholesterol levels, hypoglycemia and increased edema risk [22]. MBT1805 reduced the peak blood glucose with a smaller amplitude, and the hypoglycemic amplitude from 30 to 180 min was higher than that of rosiglitazone.
Serum biochemistry analysis revealed that MBT1805 may significantly reduce TG. Although rosiglitazone significantly lowered serum TC, TG and FFAS (p<0.001), marked elevation of hepatic TC and TG was observed (p<0.05). It has been reported that PPARγ gene activation may stimulate genes that favor triglycerides storage, thus lowering circulating free acid concentration [23]. Liver is a well characterized lipid storage organ, and PPARγ activation enhances hepatic TC and TG levels. Increased FFA uptake and β-oxidation in the liver may account for reducing circulating levels. Elafibranor, PPARα/δ agonist, has also been shown to significantly lower hepatic metabolic parameters, including hepatic TG and TC in db/db mice fed on Methionine-and Choline-Deficient (MCD) diet [24]. We suppose that hepatic TC may also reduce if we extend the administration time.
Conclusions
PPARs have been proven as viable targets for treatment of metabolic related disease. MBT1805, a novel pan-PPARs agonist, has similar activation effects on PPARα, PPARγ and PPARδ. Its EC50 values are 8.455μM, 11.94μM and11.15μM, respectively, and it is a balanced agonist of 3 isotypes. In db/db mice, MBT1805 exhibited mild hypoglycemic and hypolipidemic effects without bodyweight gain and hepatocyte damage. Our findings highlight MBT1805 as a potential treatment for diabetes.
Availability of Data and Materials
The database used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors’ Contributions
WC finished the animal experiment and drafted the manuscript with the help of SY. JXY and ZBH supplied the experimental drugs. JQL and NJQ were contributed to study design and supplied the financial support. All authors read and approved the final manuscript.
Ethics Approval and Consent to Participate
The animal experiment was approved by the Animal Care and Use Committee of the First Hospital of Jilin University (Jilin, China).
References
- Quintanilla Rodriguez BS, Correa R Rosiglitazone. StatPearls. Treasure Island (FL): StatPearls Publishing Copyright© 2020, StatPearls Publishing LLC. 2020.
- Consoli A, Formoso G. Do thiazolidinediones still have a role in treatment of type 2 diabetes mellitus? Diabetes, obesity & metabolism. 2013; 15: 967-977.
- Mirza AZ, Althagafi II, Shamshad H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. European journal of medicinal chemistry. 2019; 166: 502-513.
- Takada I, Makishima M. Peroxisome proliferator-activated receptor agonists and antagonists: a patent review (2014-present). Expert opinion on therapeutic patents. 2020; 30: 1-13.
- Im SS, Kim MY, Kwon SK, Kim TH, Bae JS, Kim H, et al. Peroxisome proliferator-activated receptor {alpha} is responsible for the up-regulation of hepatic glucose-6-phosphatase gene expression in fasting and db/db Mice. The Journal of biological chemistry. 2011; 286: 1157-1164.
- Dubois V, Eeckhoute J, Lefebvre P, Staels B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. The Journal of clinical investigation. 2017; 127: 1202-1214.
- Holst D, Luquet S, Nogueira V, Kristiansen K, Leverve X, Grimaldi PA. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim Biophys Acta. 2003; 1633: 43-50.
- Monsalve FA, Pyarasani RD, Delgado-Lopez F, Moore-Carrasco R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediators of inflammation. 2013; 2013: 549627.
- Liu S, Hatano B, Zhao M, Yen CC, Kang K, Reilly SM, et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. The Journal of biological chemistry. 2011; 286: 1237-1247.
- Krysiak R, G-D A, Okopien B. Hemostatic effects of bezafibrate and ω-3 fatty acids in isolated hypertriglyceridemic patients. Pharmacol Rep. 2011; 63: 763-771.
- Tarantino N, Santoro F, Correale M, De Gennaro L, Romano S, Di Biase M, et al. Fenofibrate and Dyslipidemia: Still a Place in Therapy? Drugs. 2018; 78: 1289-1296.
- Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: A critical review. Bioorganic chemistry. 2018; 77: 548-567.
- Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, Tan NS. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. International journal of molecular sciences. 2019; 20: 5055.
- Neelankal John A, Ram R, Jiang FX. RNA-Seq Analysis of Islets to Characterize the Dedifferentiation in Type 2 Diabetes Model Mice db/db. Endocrine pathology. 2018; 29: 207-221.
- Guest PC, Rahmoune H. Characterization of the db/db Mouse Model of Type 2 Diabetes. Methods in molecular biology. 2019; 1916: 195-201.
- Li Z, Liao C, Ko BC, Shan S, Tong EH, Yin Z, et al. Design, synthesis, and evaluation of a new class of noncyclic 1,3-dicarbonyl compounds as PPARalpha selective activators. Bioorganic & medicinal chemistry letters. 2004; 14: 3507-3511.
- Kintscher U, Law RE. PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. American journal of physiology Endocrinology and metabolism. 2005; 288: E287-E291.
- Chaput E, Saladin R, Silvestre M, Edgar AD. Fenofibrate and rosiglitazone lower serum triglycerides with opposing effects on body weight. Biochemical and biophysical research communications. 2000; 271: 445-450.
- Shim WS, Do MY, Kim SK, Kim HJ, Hur KY, Kang ES, et al. The long-term effects of rosiglitazone on serum lipid concentrations and body weight. Clinical endocrinology. 2006; 65: 453-459.
- Feng L, Luo H, Xu Z, Yang Z, Du G, Zhang Y, et al. Bavachinin, as a novel natural pan-PPAR agonist, exhibits unique synergistic effects with synthetic PPAR-gamma and PPAR-alpha agonists on carbohydrate and lipid metabolism in db/db and diet-induced obese mice. Diabetologia. 2016; 59: 1276-1286.
- Vlckova V, Cornelius V, Kasliwal R, Wilton L, Shakir SA. Hypoglycaemia with oral antidiabetic drugs: results from prescription-event monitoring cohorts of rosiglitazone, pioglitazone, nateglinide and repaglinide. Drug safety. 2009; 32: 409-418.
- Lu Y, Ma D, Xu W, Shao S, Yu X. Effect and cardiovascular safety of adding rosiglitazone to insulin therapy in type 2 diabetes: A meta-analysis. Journal of diabetes investigation. 2015; 6: 78-86.
- Moore GB, Chapman H, Holder JC, Lister CA, Piercy V, Smith SA, et al. Differential regulation of adipocytokine mRNAs by rosiglitazone in db/db mice. Biochemical and biophysical research communications. 2001; 286: 735-741.
- Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013; 58: 1941-1952.