
Abstract
A pseudopheochromocytoma is a condition where symptoms of a pheochromocytoma are present with elevated catecholamines and no tumour is identified. Causes may include autonomic epilepsy, baroreflex failure, anxiety/ panic disorders, drugs and adrenomedullary hyperplasia. A case report of a 43 year old female with frequent palpitations, sweating, headaches and extremely labile blood pressure is presented. Plasma and urine catecholamines were elevated and did not suppress with clonidine. No tumour was identified and an EEG demonstrated autonomic epilepsy. Anti-epileptic medication resulted in complete suppression of signs and symptoms and the patient remained well after 15 years of follow up.
Introduction
Modestly elevated catecholamines are a common finding in the work up of patients with hypertension without a phaeochromcytoma being identified. This ill-defined entity is termed pseudopheochromocytoma [1]. We wish to report an extremely rare cause of this condition namely autonomic epilepsy.
Case Presentation
The patient was a 43-year-old Caucasian woman who was a registered nurse. Her presenting symptoms were frequent paroxysms of headache, sweating and palpitations associated with dizziness, tiredness, nausea and blurred vision. In her family history her father had type 2 diabetes and her mother osteoporosis. There was no history to suggest multiple endocrine neoplasia. The patient had long standing depression for the last 10 years controlled with stable doses of molipaxin 50 mg and trifluoperazine hydrochloride 20 mg. During 2 of these paroxysms her blood pressure (BP) was 191/133 mmHg and 188/141 mmHg respectively. After cessation of the paroxysm her BP was 80/50 mmHg with a pulse of 50 beats/min with symptoms of postural hypotension. The rest of the examination was unremarkable. Several 24 ambulatory BP monitors confirmed the paroxysms of severe hypertension followed by hypotension.
All the following tests were performed during her first admission. The full blood count, urea, creatinine and electrolytes, thyroid function, cortisol, ACTH, Synacten test, PTH, ionized calcium, baroreceptor sensitivity, ECG and stress ECG were all normal. Tests for LH, FSH and estrogen showed no signs of menopause.
Blood and urine tests for catecholamines and a clonidine suppression test were performed (Tables 1 & 2). These results were suggestive of an epinephrine producing pheochromocytoma.
![]()
Result
Upper reference limits
Total 24 hour urine metanephrine x 2
40, 2 and 38.4 μmol/24 hr
< 5 umol/24 hr
Random norepinephrine x 2
433 and 349 pmol/L
450-2490 pmol/L
Random epinephrine x 2
1310 and 1070 pmol/L
20-460 pmol/L
Table 1: Plasma and urine tests for catecholamines.
![]()
Time
Epinephrine
Norepinephrine
Dopamine
60 min
1578 pmol/L
335 pmol/L
<30 pmol/l
120 min
1545 pmol/L
276 pmol/l
<30 pmol/l
180 min
1742 pmol/L
309 pmol/L
<30 pmol/l
Table 2: Clonidine suppression test.
A MIBG scan showed questionable uptake in the left paravertebral area but the MRI of this area, neck, thorax, head, kidney, adrenal, and abdomen was normal.
An EEG was performed and revealed features in keeping with diencephalic or autonomic epilepsy. A diagnosis of pseudopheochromocytoma due to automonic epilepsy was made. The patient was treated with topiramate 50 mg daily and oxycarbazipine 600 mg daily with complete resolution of symptoms. Her antidepressants were continued. The patient has remained stable for 15 years on treatment. No repeat metanephrine testing was done after institution of treatment.
Discussion
Pheochromocytomas and Paragangliomas (PPGLs) are chromaffin cell tumours that arise from the adrenalmedulla in 80-85% of patients and from extra-adrenal sympathetic tissue of abdomen, pelvis and chest [2]. Typically they present with paroxysms of sweating, headache, palpitations, and labile BP with both severe hypertension and hypotension due to excessive release of catecholamines.
However there is a group of patients who have similar paroxysms with modestly elevated catecholamines (usually no more that 4x the upper limit of normal) in whom no tumour is identified (Figure 1) [1,3]. This is termed pseudophaechromocytoma, and remains a diagnosis of exclusion. In many cases the cause is unknown [4] but in 40% it is due to attacks of anxiety with sympathetic over activity [5]. Other causes are listed (Table 3) [3,6-9].
![]()
Anxiety, panic attacks
Hyperthyroidism
Migraine headaches
Hypertensive encephalopathy
Coronary insufficiency
Reno vascular hypertension
Central nervous system lesions such as stroke, tumor, bleed
Seizures – autonomic or diencephalic epilepsy
Carcinoid
Monamine oxidase inhibitor, clozapine, tricyclic antidepressant
Baroreflexdysfunction
Factitious hypertension – ingestion of sympathomimetics (cocaine, amphetamines, ephedrine)
Adrenomedullary hyperplasia
Table 3: Causes of pseudopheochromocytoma [3,5-10].
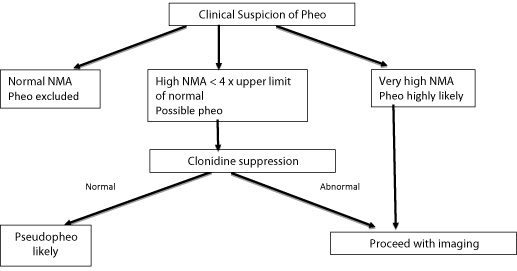
Figure 1: Proposed approach to patient with symptoms of pheochromocytoma.
In this case report the patient presented with typical symptoms of a pheochromocytoma with elevated total urinary metanephrines and plasma epinephrine. However no definitive tumour was demonstrated on MIBG and MRI scanning making the diagnosis of pseudopheochromocytoma likely. Sharabi et al. showed that plasma epinephrine levels and not norepinephrine levels are increased in this condition [1].
A clonidine suppression test was performed, which showed no suppression of the elevated plasma epinephrine suggesting anxiety/panic attacks were an unlikely aetiology [2]. Most causes of pseudopheochromocytoma (Table 3) were considered unlikely or excluded. Her antidepressant drugs were thought not to be implicated because she had been on stable doses for many years without symptoms. Adrenomedullary hyperplasia or renal cysts can cause isolated hyper epinephrinaemia without pheochromocytoma, [11] but the MRI studies did not demonstrate these abnormalities.
Autonomic or diencephalic epilepsy could not be excluded and the patient was referred to a neurologist for EEG examination. This showed typical features of autonomic epilepsy. Further proof supporting the diagnosis was the complete response to anti-epileptic medication and the extended period of follow of 15 years. The only atypical feature of this case was the failure to suppress catecholamines by clonidine as previously described by Metz et al. in autonomic epilepsy [6].
Conclusion
We suggest testing for autonomic or diencephalic epilepsy in cases of unexplained pseudopheochromocytoma, as this is a highly treatable condition with excellent outcome.
References
- Sharabi Y, Goldstein DS, Bentho O, Saleem A, Pechnik S, Geraci MF, et al. Sympathoadrenal function in patients with paroxysmal hypertension: pseudopheochromocytoma. J Hypertens. 2007; 25: 2286-2295.
- Van Berkel A, Lenders JW, Timmers HJ. Diagnosis of endocrine disease: Biochemical diagnosis of phaeochromocytoma and paraganglioma. Eur J Endocrinol. 2014; 170: 109-119.
- Kaplan N. Pheochromocytoma (with incidental preface about incidental renal masses). Kaplan N, Flynn JT. In: Kaplan’s Clinical Hypertension (9th Edn), Lippincott, Williams, Wilkins, Philadelphia. 2009; 12: 389-409.
- Kuchel O. Pseudopheochromocytoma. Hypertension. 1985; 7: 151-158.
- Fogarty J, Engel CC, Russo J, Simon G, Katon W. Hypertension and pheochromocytoma testing. The association with anxiety disorders. Arch Fam Med. 1994; 3: 55-60.
- Metz SA, Halter JB, Porte D Jr, Robertson RP. Autonomic epilepsy: clonidine blockade of paroxysmal catecholamine release and flushing. Ann Intern Med. 1978; 88: 189-193.
- Mann SJ. Severe paroxysmal hypertension (pseudopheochromocytoma): understanding the cause and the treatment. Arch Int Med. 1999; 159: 670- 674.
- Zar T, Peixoto AJ. Paroxysmal hypertension due to baroreflex failure. Kidney Int. 2008; 74: 126-131.
- Rudy FR, Bates RD, Cimorelli AJ, Hill GS, Engelman K. Adrenal medullary hyperplasia: a clinicopathologic study of four cases. Hum Pathol. 1980; 11: 650-657.
- Kothari SS. When epilepsy masquerades as heart disease. Awareness is key to avoiding misdiagnosis. Postgrad Med. 1990; 88: 167, 170-171.
- Streeten DH, Anderson GH Jr, Lebowitz M, Speller PJ. Primary hyperepinephrinemia in patients without pheochromocytoma. Arch Intern Med. 1990; 150: 1528-1533.