
Editorial
Austin Renal Dis. 2016; 1(1):1004.
Podocytes Globotriaosylceramide Content as a Potential Biomarker for Early Monitoring the Efficacy of Enzyme Replacement Therapy in Fabry Disease
Pozdzik A¹* and Brocheriou I2,3
¹Nephrology and Dialysis Department, Centre Hospitalier Chrétien - Clinique Saint-Joseph, Liège, Belgium
²Pathology Department, AP-HP Pitié Hospital, Paris, France
³UPMC Univ-Paris 6, Paris, France
*Corresponding author: Agnieszka POZDZIK, Nephrology and Dialysis Department, Centre Hospitalier Chrétien - Clinique Saint-Joseph, Rue de Hesbaye, 75, B-4000 Liège, Belgium
Received: October 03, 2016; Accepted: October 06, 2016; Published: October 10, 2016
Editorial
Fabry Disease (FD) is an inherited X-linked lysosomial storage disorder secondary to the mutation of alpha-galactosidase (αGAL) gene [1]. The αGAL gene impairment results in various defects of αGAL enzymatic activity ranging from slight decrease of the amount of enzyme to its complete absence. FD is mainly described in men. However, women could be also affected, sometimes, with the same degree of severity. This can be explained by sporadic “de novo” mutations [2,3] of αGAL or by the inactivation of unaltered X-chromosome in heterozygous carriers. Therefore, the careful screening for this disease in all suspected female patients without any familial context is also strongly suggested (Figure 1) [4].
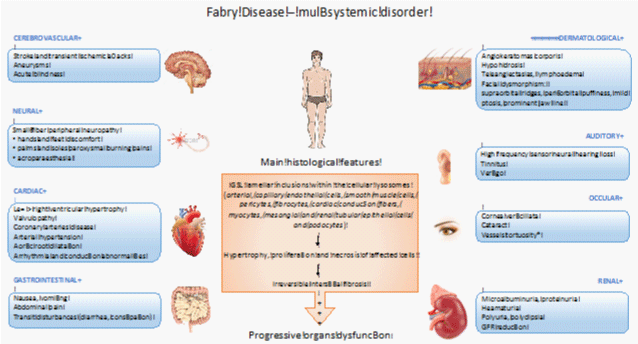
Figure 1: The main clinical and biological characteristics of Fabry disease.
GFR: Glomerular Filtration Rate; *: Indicate that this feature is associated with worse prognosis.
Despite the fact that the endorsement of the diagnosis is easy, FD is still under-diagnosed, probably because of very heterogeneous presentations (Figure 1). In men suspected of FD, the assessment of αGAL enzymatic activity could be performed in plasma, in circulating leukocytes and skin fibroblasts [5]. However, in women, additional genetic testing for αGAL mutation is required [5].
The pathophysiology of FD is only poorly understood until now. It is well recognized that the disease starts by lysosomal accumulation of αGAL substrates within the cells of different organs. Intracellular globotriaosylceramide (GL3) and related neutral glycosphingolipids inclusions induce adaptive hypertrophy, proliferation and lastly necrosis of affected cells. Continuous increase in glycosphingolipids leads finally to tissue interstitial fibrosis resulting in several organs dysfunction (Figure 1).
It has been reported that early deficient Enzyme Replacement Therapy (ERT) could impede the progression of GL3 related tissue damages to devastating stage. However, long-term benefits have not been established yet [6-8]. ERT [agalactosidase alpha (Replagal ®, Shire HGT, Inc) and agalactosidase beta (Febrazyme ®, Genzyme- Sanofi Corp)] exists from more than 10 years now and is safe and well tolerated. Recently, the European recommendations for ERT have been published [9]. However, there is still no consensus about the start of ERT, for how long and when to stop it [6]. This could be explained at least by 1) a huge intra- and interfamilial heterogeneity of clinical and biological FD presentations even in the presence of the same mutation, 2) sporadic “de novo” GLA3 mutations in the patients without any familial evidence of FD (delayed diagnosis) and 3) the absence of robust biomarker(s) of FD disease activity.
All these features strongly support the vigilant individual assessment of each suspected patient and incite to personalized treatment based on the disease activity. Consequently, they denote the urgent need for new biomarker(s) for adequate assessment of FD activity and severity as well as for long follow-up monitoring of ERT efficacy [9].
To find the new robust biomarker(s) that will be reliable for FD activity, with the tools currently used in clinical practice and ERT, it is entirely reasonable and very tempting to focus on renal tissue changes. Indeed, renal involvement precedes in general other organs injury and progressive kidney disease is associated with cardiovascular morbidity and mortality.
Fabry nephropathy is well described [10,11]. It is characterized by GL3 intracellular inclusions within the glomerulus in endothelial, epithelial and mesangial cells as well as in podocytes. Sustained increase in GL3 depositions induces progressive podocytopathies resulting in intracellular vacuolization associated with focal and, later, global foot process effacement and finally total loss of podocytes. The same GL3 inclusions have also been described in distal, and, less frequently, in proximal tubular epithelial cells. Within the arterioles and artery walls, myocytes and endothelial cells, GL3 inclusions are associated with the hyaline depositions. In the early stage, these changes are asymptomatic but, if left untreated, they can lead to nonspecific biological manifestations such as (micro) albuminiuria or hematuria associated or not with Glomerular Filtration Rate (GFR) decline [5].
New and attractive tools in the every-day clinical management of FD patients under ERT have been highlighted during the “3th Fabry disease nephrology experts meeting” happened in Bergen, Norway in September 2016. Firstly, the podocyturia that could reflect podocytes regeneration (difficult to prove at this time) and secondly, the evaluation of lysosomial metabolite lyso-GL3 plasma level. However, the cut-off of lyso-GL3 and related FD activity was debated. Besides these both biomarkers, the experts highlighted the crucial importance of the baseline and follow-up kidney biopsy analyses in FD patients. The classical assessment of GL3 content by conventional optical microscope using score proposed by the International Scoring Group of Fabry Nephropathy estimates the proportion of GL3 inclusions within the podocytes. Indeed, previous studies have reported that podocytes GL3 depositions evaluated by this method were only poorly affected by ERT contrasting with its rapid clearance form mesangial and endothelial cells. Therefore, podocytes GL3 deposition clearance has been suggested to be a very interesting target for ERT efficacy [12]. Consequently, the question of ERT duration and doses followed this purpose with the risk of potential serious adverse effects such as anaphylactic reactions or allo-immunisation with potential risk of enzyme inefficacy. Morphological data of GL3 related podocytopathy has been underlined than as a useful and interesting tool to manage ERT in FD patients and completed the list of 3 new useful biomarkers in the decision for initiation of ERT.
New electron microscopic stereological method estimating the average volume of GL3 depositions and volume of podocytes demonstrated that ERT induces a very significant deposition removal, reduce the podocytes volume and foot process after at least 11 months of ERT [13]. Besides the evidence that estimation of ratio vo/vol GL3 content/podocytes is invasive, assessment requiring the electron microscopy is, without any doubts, a time-consuming procedure. However, this method also evaluates the exocytosis of GL3 by podocytes, bringing new information on the mechanism of GL3 deposition removal induced by ERT. Consequently, these preliminary data reporting the usefulness of stereological measurement of GL3 podocytes content as a new biomarker for FD patient’s need to be further confirmed in larger cohort.
To do this the standardized time schedule of kidney tissue biopsy, the processing and the storage of kidney tissue samples needs to be established. Therefore, the patients’ registries as well as international collaborative networks of reference centers are highly encouraged to incite collaboration, to establish a comprehensive care plan and to empower the number of followed patients [14,15].
Summarizing, repeated kidney biopsy is strongly encouraged as an opportunity to better estimating the balance efficacy-side effects in patients currently under ERT and to improve our understanding of initiation and progression mechanisms, at least, of renal involvement during Fabry disease. The precise GL3 deposition clearance evaluation is actually possible in specialized centers. Prospective clinical trials testing ERT and new drugs using the new electron microscopic stereological tridimensional measurement of podocytes GL3 content will provided a new insight on induction, progression and reversibility of podocytopathy in FD.
References
- Tuttolomondo A, Pecoraro R, Simonetta I, Miceli S, Pinto A, Licata G. Anderson-Fabry disease: a multiorgan disease. Curr Pharm Des. 2013; 19: 5974-5996.
- Pisani A, Bruzzese D, Sabbatini M, Spinelli L, Imbriaco M, Riccio E. Switch to agalsidase alfa after shortage of agalsidase beta in Fabry disease: a systematic review and meta-analysis of the literature. Genet Med. 2016.
- Zizzo C, Monte I, Pisani A, Fatuzzo P, Riccio E, Rodolico MS, et al. Molecular and clinical studies in five index cases with novel mutations in the GLA gene. Gene. 2016; 578: 100-104.
- Terryn W, Cochat P, Froissart R, Ortiz A, Pirson Y, Poppe B, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant. 2013; 28: 505-517.
- Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, et al. Fabry disease: a review of current management strategies. QJM. 2010; 103: 641-659.
- El Dib R, Gomaa H, Carvalho RP, Camargo SE, Bazan R, Barretti P, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016; 7: CD006663.
- Hughes DA. Fabry disease: will markers of early disease enable early treatment and better outcomes? Curr Opin Cardiol. 2016; 31: 434-439.
- Ito S, Ogura M, Kamei K, Matsuoka K, Warnock DG. Significant improvement in Fabry disease podocytopathy after 3 years of treatment with agalsidase beta. Pediatr Nephrol. 2016; 31: 1369-1373.
- Biegstraaten M, Arngrimsson R, Barbey F, Boks L, Cecchi F, Deegan PB, et al. Recommendations for initiation and cessation of enzyme replacement therapy in patients with Fabry disease: the European Fabry Working Group consensus document. Orphanet J Rare Dis. 2015; 10: 36.
- Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002; S134-138.
- Najafian B, Svarstad E, Bostad L, Gubler MC, Tondel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011; 79: 663-670.
- Skrunes R, Svarstad E, Kampevold Larsen K, Leh S, Tondel C. Reaccumulation of globotriaosylceramide in podocytes after agalsidase dose reduction in young Fabry patients. Nephrol Dial Transplant. 2016.
- Najafian B, Tondel C, Svarstad E, Sokolovkiy A, Smith K, Mauer M. One Year of Enzyme Replacement Therapy Reduces Globotriaosylceramide Inclusions in Podocytes in Male Adult Patients with Fabry Disease. PLoS One. 2016; 11: e0152812.
- Parker S. The pooling of manpower and resources through the establishment of European reference networks and rare disease patient registries is a necessary area of collaboration for rare renal disorders. Nephrol Dial Transplant. 2014; 9-14.
- Hopkin RJ, Jefferies JL, Laney DA, Lawson VH, Mauer M, Taylor MR, et al. The management and treatment of children with Fabry disease: A United States-based perspective. Mol Genet Metab. 2016; 117: 104-113.