
Research Article
Austin J Autism & Relat Disabil. 2016; 2(3): 1023.
Mutation Screening and Copy Number Detection of NRXN1 in Chinese Han Patients with Autism
Liu W-W¹, Wen Z1,2, Zhang L-N¹, Gong X-H³, Wang H-Y³ and Du Y-S1,2*
¹Department of Child and Adolescent Psychiatry, Shanghai Mental Health Center, China
²School of Medicine, Shanghai Jiao Tong University, China
³The MOE Key Laboratory of Contemporary Anthropology and State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, China
*Corresponding author:Ya-Song Du, Shanghai Jiatong University Affiliated Mental Health Center Child and Adolescent Psychiatry, Shanghai, China
Received: April 19, 2016; Accepted: June 21, 2016; Published: June 22, 2016
Abstract
Objective: To explore whether the coding and regulatory regions in the Neurexin1 (NRXN1) gene are varied in Han Chinese children with autism, and to predict the possible function of these mutations.
Methods: 285 children from nuclear families diagnosed with autism, according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth edition (DSM-IV), and 384 healthy students as controls were enrolled. The coding regions and the regulatory regions, including the promoter region and the 5’ and 3’ Untranslated Regions (UTRs) in the NRXN1were screened for mutations by Sanger sequencing. Copy number variations of NRXN1 were detected using multiplex fluorescence competitive PCR. Chi-squared test was used to compare the allele and genotype frequencies between cases and controls.
Results: 8 missense mutations and 2 SNPs were identified in the NRXN1 gene. Among them, 5missense mutations were found in NRXN1-α, including p.G65S (c.193G>A), p.P159S (c.475C>T), p.D206H (c.616G>C), p.I794T (c.2381T>C) and p.I1086V (c.3202A>G); and 3 missense mutations were found in NRXN1-β, including p.S14L (c.41C>T), p.R345X (c.1033C>T) and p.R345Q (c.1034G>A). 7 of them were novel and only p.S14L has been previously reported. p.P159S and p.D206H were de novo mutations, and the remainders were maternally inherited. Two SNPs (rs13422484 and rs3732049) were found in the 5’UTR region of the NRXN1. The genotype frequencies were not significant different between cases and controls (P = 0.79 and 0.053 respectively). Four sequence changes (-1576A>G, -1192delA, -331G>A and -154>T) were detected in the promotor region of NRXN1-β and were absent in control subjects.
Conclusion: Our results suggest that NRXN1 may be a susceptibility gene for Han Chinese children with autism.
Background
Autism Spectrum Disorder (ASD) is neurodevelopmental disorders which prevalence has steadily increased over the past decade [1]. According to the 5th edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5), the core symptoms of ASD are persistent deficits in social communication and social interactions and restricted, repetitive patterns of behavior, interests, or activities ASD symptoms are often accompanied by mood swings, irritability, paresthesia, hyperacusis, visual avoidance, aggression, self-injurious behavior and other abnormal symptoms. These disturbances are not better explained by intellectual disability or global developmental delay [2]. Genetic studies indicate that Neurexin-Neuroligin (NRXNNLGN) pathway genes contribute susceptibility to ASD [3,4]. As synaptic cell-adhesion molecules, neurexins and neuroligins mediate trans-synaptic signaling, and are involved in shaping neural network properties by specifying synaptic functions [4]. In humans, variations in NRXN or NLGN genes are implicated in ASD and other mental development diseases. Therefore, NRXNs and NLGNs are kernel components of the molecular machinery that regulates synaptic transmission and approves neural networks to produces complex signals.
Vertebrates have three large neurexin genes, each of which expresses longer a-neurexins and shorter b-neurexins [5]. a-Neurexins bind neurexophilin, while b-Neurexins bind neuroligins to form an intercellular junction [6]. Epilepsy occurs in patients with ASD has fueled speculation that autism may be caused by an imbalance of excitatory vs. inhibitory synaptic processes [7]. Protein complexes are associated with NLGNs and NRXNs at excitatory and inhibitory synapses [8]. In postsynaptic inhibitory synapses, both α-NRXN and β-NRXN can interact with dystroglycan. At presynaptic terminals, both α- and β-neurexins can directly interact with a complex containing CASK/Veli/Mint-1, and trigger the presynaptic vesicle exocytosis machinery. In addition, neurexins can also directly bind to synaptotagmins [9].
Indeed, recent studies have identified mutations in the genes encoding Nrxns and Nlgns as a cause for ASD [10-13]. Two different deletions of Nrxn1 have been observed in families with schizophrenia, indicating that there is a continuum of disorders that involves dysfunctions in synaptic cell adhesion that manifests in different ways [14]. Conversely, different molecular changes may produce a similar syndrome, as exemplified by different mutations associated with ASDs. The relationship between the Nrxn/Nlgn synaptic celladhesion complex and ASDs is scarce. On one hand, many mutations observed in familial ASD are clearly not polymorphisms but are deleterious, as evidenced by the effect of these mutations on the structure or expression of the corresponding genes, and by the severe autism-like phenotypes observed in Nlgn3 and Nlgn4 mutant mice. On the other hand, the nonlinear genotype/phenotype relationship in humans, evident from the 70–80% heritability and from the occasional presence of mutations in non-symptomatic individuals, requires explanation. Elucidating underlying mechanisms for this incomplete genotype/phenotype relationship is a promising avenue into the genesis of autism.
Furthermore, in addition to the link of Nrxn1 mutations with schizophrenia, linkage studies have connected Nrxn3 to different types of addiction. Friedman’s group (2006) identified a de novo heterozygous deletion in the NRXN-1a promoter and exons 1-5 in a 7 year-old boy with cognitive impairment, autistic features, vertebral anomalies, and mild facial dysmorphism, offering initial evidence for neurexin-1 in neurodevelopment disorders [15]. Subsequently, more than ten studies confirmed the association between NRXN1 genetic variants and ASD. Coding regions and associated splice junctions of three NRXN-1b genes were scanned, and two putative missense structural variants were identified in the NRXN-1b gene in four Caucasian patients with autism [10]. Szatmari and coworkers, using a whole-genome approach, identified a de novo heterozygous deletion eliminating exons of NRXN-1a and NRXN-1b in two siblings with typical autism and language regression [16]. Kim’s group reported two missense changes in conserved residues of the NRXN-1a leader sequence and an Epidermal Growth Factor (EGF) -like domain respectively6. The NRXN1 deletions associated with autism have also been studied in patients with schizophrenia [17], who share some genetic risk factors with autism. Rujescu and colleagues examined NRXN-1, NRXN-2 and NRXN-3 genes using microassay analysis, and reported a statistically significant association of NRXN -1 deletions with the risk of schizophrenia [18].
Only one study suggested a susceptibility of NRXN1 to ASD in a Chinese population and 22 variants in the NRXN1 coding regions [19], including 7 missense variants, 3 deletions, and 12 synonymous mutations have been identified. Among them, 7 missense mutations were not reported in the dbSNP database. However, few studies exist to indicate a significant association of these mutations with autism risk. There was a statistically significant association of NRXN1 SNP P300P (rs2303298) with the risk of autism (P = 3.45×-00-6; OR = 2.152, 95%CI: 1.559-2.970).
Possibly, because of the nature of their function, mutations in genes encoding Nrxns constitute “hot spots” for human cognitive diseases. Neurexins and neuroligins maintain functional excitatory synapses and inhibitory balance of essential molecules and ~1% of children with autism have mutations in these genes. Expressed in the presynaptic membrane neurexin, this trans-synaptic complex is a necessary component of synaptic formation and effective exchange of information between synapses. Thus, neurexin proteins may play an important role in synaptic membrane adhesive proteins, bridging synapses to promote synaptic signal transmission. Peculiarities in this process may be a pathological basis of autism.
To explore whether the NRXN1 gene is varied in Han Chinese children with autism, and whether NRXN1 mutations affect functions of the encoded protein. In the present study, we screened a cohort of 285 ASD patients from the Outpatient Department of the Child and Adolescent Psychiatry, Shanghai Mental Health Center and 384 healthy controls recruited from students in Fudan University mutations in the entire coding region and the regulatory region, including the promoter region and 5’ or 3’ UTRs in NRXN1(a- NRXN1,b-NRXN1). We also examined copy number variations of NRXN1 in these study subjects.
Method
Ethics statement
The study was approved by the Ethics Committee of Shanghai Mental Health Center and the Ethics Committee of the School of Life Sciences, Fudan University. The methods in this study were carried out in accordance with the approved guidelines which are in supplementary. We identify the Ethics committee approving the experiments, and include with our submission a statement confirming that informed consent was obtained from all subjects.
Study subjects: 285 patients with ASD from Shanghai Mental Health Center and 384 healthy controls were recruited from students in Fudan University. The detailed sample information is in accordance with the publish before.
DNA sequencing
The reference sequence of the NRXN1 gene was obtained from the UCSC Genome Browser (NRXN1-α, NM_001135659.1; NRXN1-β, NM_138735.2). The promoter region, twenty-four exons including the 5’/3’ UTRs and the Open Reading Frame (ORF) were sequenced. The methods of PCR, extracting DNA from blood followed those described previously. The Mutation Surveyor software was used to read the sequencing results. The identified variants were confirmed in the NCBI SNP database as known or novel. A novel variant was described using the mutation nomenclature from the HGVS. The first nucleotide of the translational start site is designated as +1 and the nucleotide upstream from that was -1.
Detection of NRXN1 copy number variations
The method to measure The NRXN1 copy number and principle of multiplex fluorescence competitive PCR as described by Du’s group. Three fragments (exon 5 and the intron 5, intron 18, and exon 22) in NRXN1 were assayed. The parameters of PCR reaction were same with the description before [20]. The sample/competitive (S/C) peak ratio was calculated for exon 5, intron 5, intron 18, and exon 22 fragments of NRXN1 and three reference genes (POP1, RPP14, and TBX15) and the S/C ratio for each target fragment was first normalized to three reference genes respectively.
Statistical analysis
SPSS for Windows (version 17.0; SPSS Inc., Chicago, IL) was used for all statistical analysis. Chi-squared test was used to evaluate the association of 2 common SNPs with the disease. Multiple comparisons were corrected using the Bonferroni method.
Result
Eight missense mutations were identified in the coding regions of NRXN1. Of which, 5 mutations were identified in NRXN1-α: p.G65S (c.193G>A), p.P159S (c.475C>T), p.D206H (c.616G>C), p.I794T (c.2381T>C) and p.I1086V (c.3202A>G); and 3 mutations were identified in NRXN1-β: p.S14L (c.41C>T), p.R345X (c.1033C>T) and p.R345Q (c.1034G>A). Among these mutations, 7 were novel and p.S14L was previously reported. p.P159S and p.D206H were de novo mutations, and the others were maternally inherited (Figure 1A , 1B and Table 1).
![]()
Amino acid change
Sequence change
Isoform
Genomic position
Target exon
Regions
Predicted function
Transmission
NRXN1-α
p.G65S
c.193G>A
NM_001135659.1
Chr2: 51255219
2
Laminin G-like 1
Benign
NRXN1-α
p.P159S
c.475C>T
NM_001135659.1
Chr2: 51254937
2
Laminin G-like 1
Possibly damaging
de novo
NRXN1-α
p.D206H
c.616G>C
NM_001135659.1
Chr2: 51254796
2
Laminin G-like 1
de novo
NRXN1-α
p.I794T
c.2381T>C
NM_001135659.1
Chr2: 50758451
12
Laminin G-like 4
Benign
Maternally inherited
NRXN1-α
p.I1086V
c.3202A>G
NM_001135659.1
Chr2: 50699598
17
EGF-like 3
Benign
Maternally inherited
NRXN1-β
p.S14L
c.41C>T
NM_138735.2
Chr2: 50427551
1
Signal peptide
Benign
Maternally inherited
p.R345X
c.1033C>T
NM_138735.2
Chr2: 50002882
6
between Topological domain and Transmembrane
Maternally inherited
NRXN1-β
p.R345Q
c.1034G>A
NM_138735.2
Chr2: 50002881
6
between Topological domain and Transmembrane
Benign
Maternally inherited
Table 1: Missense mutations in the NRXN1 gene identified in this study.
Two SNPs rs13422484 and rs3732049 were found in the 5’UTR region of the NRXN1 gene and the genotype frequencies were not significant different between cases and controls (P = 0.79 and 0.053 respectively Table 2). Four sequence changes (-1576A>G, -1192delA, -331G>A and -154>T) were detected in the promoter region of NRXN1-β and were absent in control subjects.
![]()
Genotype
Case n (%)
Control n (%)
Pa
Pb
Pc
rs3732049
GG
GA
AA
222 (77.1)
62 (21.5)
4 (1.4)
298 (79.3)
73 (19.4)
5 (1.3)
0.79
0.85
0.395
rs13422484
TT
CT
CC
78 (27.1)
142 (49.3)
68 (23.6)
135 (35.9)
163 (43.3)
78 (20.7)
0.05
0.07
0.025
aChi-squared test for association between the SNP and ASD. bChi-squared test for Hardy-Weinburg equilibrium. cBonferroni method for multiple testings.
Table 2: Association analysis of 2 SNPS in the NRXN1-β gene with ASD.
The detected copy numbers of three fragments of NRXN1 varied between 1.53 and 2.51 in 284 samples except for 1.03 in one individual, which indicated a deletion in the three fragments. This sample was then selected for validation by Sanger sequencing and Multiplex Ligation-Dependent Probe Amplification (MLPA) and a fragment of 508 kb deletion was confirmed in this sample (Figure 2). Reported sequence variations of NRXN1 in previous studies were summarized in Table 1.
Figure 2: Assays of copy number in the NRXN1 gene.
Discussion
In the present study, we investigated the role of NRXN1 in Chinese patients with ASD by comprehensively testing genetic variants including rare mutations in the coding and regulatory regions, as well as copy number variations and common SNPs. Interestingly, among coding variants in NRXN-1a, three variants (p.G65S, P159S and D206H) were identified in the predicted Laminin G-like 1 domain. One variant (p.I794T) was located in the predicted Laminin G-like 4 domain, and one variant (p.I1086V) was found in the predicted EGF-like 3 domain. Three mutations were detected in NRXN1-β, one (p.S14L) located in the predicted signal peptide domain, the other two mutations (p.R345X and p.R345Q) located between topological and transmembrane domains (Figure 1B). Two online software tools (polyphen-2: prediction of functional effects of human SNPs; and protein variation effect analyzer) were used to predict mutation influences on protein. First, NRXN1 is a conservative gene and the sites or areas of these mutations have a high degree of evolutionary conservation throughout different species by homology analysis. As the results of polyphen-2: p.R345X is damaging, p.P159S and p.D206H are possible damaging, and the others seem as benign. We speculated that variants mentioned above may modify the binding of the neurexin-neuroligin complex during neurodevelopment. Genetic mutations in NRXN1-β are proposed to disrupt synaptic alignment and neural circuitry, which may underlie pervasive psychiatric disorders such as schizophrenia and ASD.
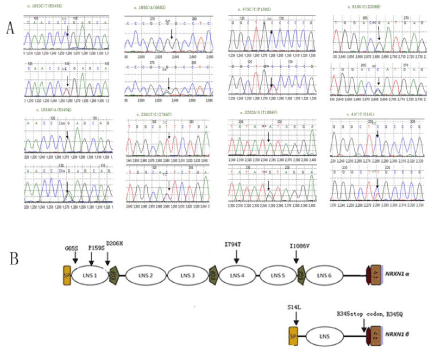
Figure 1: A. Sequence diagrams of 8 missense mutations in NRXN1 identified in this study. B. The gene structure of NRXN1 showing 8 missense mutations
identified in this study.
Unlike many other diseases, autism does not have a primary genetic etiology. Whereas many of genes discussed have altered expression, few polymorphisms have been identified. Therefore, specific genetic links remain unclear. Thus, it is essential to explore the role of epigenetic modulators in the etiology of autism. In our study, six mutations were inherited from unaffected mothers and two were de novo after analysis of available parent’s DNA, suggesting that there may be an imprinting effect on gene expression.
Most previously reported studies focused on mutations in the coding region. In this study, we identified genetic variants which may influence NRXN1 expression. Three rare mutations in the promoter region or 5’ and 3’ UTRs of NRXN1 were identified in our samples. These mutations may affect NRXN1 expression by binding with transcription factors or micro RNA, and they were predicted to be binding sites for transcription factors N1T2, ADR1 and cap using the online tool TFSEARCH. In addition, we did not find any significant association of the two SNPs in the NRXN1-β gene with ASD (Table 2). Further investigation in larger samples and including functional assays would help us to understand the role of these common polymorphisms in ASD.
In summary, we investigated the role of NRXN1 in ASD by comprehensive screening of the coding and regulatory regions of NRXN1 for mutations and detecting copy number variations of NRXN1. The purpose of our study is that the results suggest a possible role of NRXN1 mutations in autism in a Chinese Han population; however, additional large-scale studies are needed to confirm these findings.
Acknowledgement
We are grateful to the children and parents who participated in this study. This work was supported by the National Program on Key Basic Research Project (973 Program) of China (Grant No. 2010CB529602) and the Research Program of Shanghai Jiao Tong University (Grant No. YG2013ZD0503).
References
- Baio J. Prevalence of Autism Spectrum Disorders: Autism and Developmental Disabilities Monitoring Network. Surveillance Summaries. 2012; 61: 1-19.
- Association AP. The Diagnostic and Statistical Manual of Mental Disorders: DSM 5. 2013.
- Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008; 82: 199-207.
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008; 455: 903-911.
- Siidhof TC. The Synaptic Cleft and Synaptic Cell Adhesion. Synapses. 2003: 275.
- Nguyen T, Südhof TC. Binding properties of neuroligin 1 and neurexin 1beta reveal function as heterophilic cell adhesion molecules. J Biol Chem. 1997; 272: 26032-26039.
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003; 2: 255-267.
- Levinson JN, El-Husseini A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron. 2005; 48: 171-174.
- Lisé MF, El-Husseini A. The neuroligin and neurexin families: from structure to function at the synapse. Cell Mol Life Sci. 2006; 63: 1833-1849.
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, et al. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006; 409: 10-13.
- Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013; 93: 249-263.
- Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda). 2012; 2: 1665-1685.
- Yan J, Noltner K, Feng J, Li W, Schroer R, Skinner C, et al. Neurexin 1alpha structural variants associated with autism. Neurosci Lett. 2008; 438: 368-370.
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008; 320: 539-543.
- Friedman SD, Shaw DW, Artru AA, Dawson G, Petropoulos H, Dager SR. Gray and white matter brain chemistry in young children with autism. Arch Gen Psychiatry. 2006; 63: 786-794.
- Autism Genome Project C, Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007; 39: 319-328.
- Mühleisen TW, Basmanav FB, Forstner AJ, Mattheisen M, Priebe L, Herms S, et al. Resequencing and follow-up of neurexin 1 (NRXN1) in schizophrenia patients. Schizophr Res. 2011; 127: 35-40.
- Rujescu D, Ingason A, Cichon S, Pietiläinen OP, Barnes MR, Toulopoulou T, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2009; 18: 988-996.
- Liu Y, Hu Z, Xun G, Peng Y, Lu L, Xu X, et al. Mutation analysis of the NRXN1 gene in a Chinese autism cohort. J Psychiatr Res. 2012; 46: 630-634.