
Special Issue - Genetics of Autism Spectrum Disorder
Austin J Autism & Relat Disabil. 2017; 3(1): 1037.
A Case Control Study to Compare the Phenotype of Children in Lothian with Autism Spectrum Disorder and Either a Sub Microscopic Deletion or Duplication
Fairfield RE* and Clegg SK
Department of Community Child Health’s, University of Edinburgh, UK
*Corresponding author:Rachel Fairfield, Department of Community Child Health’s, University of Edinburgh, UK
Received: February 27, 2017; Accepted: April 12, 2017; Published: April 19, 2017
Abstract
Introduction: 10% of ASD cases are accounted for by genetic syndromes. There is much interest in susceptibility genes that may contribute to the remaining 90% of “idiopathic” autism. In the last 5 years array CGH has made it possible to identify sub microscopic deletions and duplications. These are either inherited or de novo and have been reported in 10-35 % of ASD cases.
Methodologies: A database of children attending the ASD clinic and the genetics clinic was combined; of these 34 patients met the inclusion criteria of age <18, a diagnosis of ASD and a sub-microscopic deletion or duplication. The genetic diagnosis, its inheritance, the family history, the peri-natal and neonatal history, phenotype and sensory issues were recorded.
Results: The odds ratios for having a deletion increased the chance of maternal bleeding, birth complications, having a lower birth weight, having a learning disability, structural anomalies and dysmorphisms. The odds ratio of a duplication having a family history of ASD was also increased.
Conclusion: Deletions were more likely to be de novo and associated with a more severe phenotype of ASD whereas the duplications were more likely to be inherited and associated with a family history of ASD.
Keywords: Autism spectrum disorder; Genetics; Submicroscopic deletion; Chromosome
Abbreviations
ASD: Autism Spectrum Disorder; Array CGH: Chromosome Array Genomic Hybridisation; CNV: Copy Number Variants (CNV); MCHAT: Modified Checklist for Autism in Toddlers; ADI-R: The Autism Diagnostic Interview – Revised (ADI–R)
Introduction
Autism Spectrum Disorder (ASD) is a neurodevelopmental disorder. It consists of a triad of symptoms that incorporate impairment in social interaction, impaired verbal and non–verbal communication skills and repetitive/stereotyped behaviour [1,2]. ASD has a male predominance and affects around 1% of the childhood population [1].
Due to the high prevalence of ASD all professionals working with young children must be “ASD aware” as early diagnosis and intervention is believed to be advantageous, although this is difficult to prove [3]. When concerns of a possible ASD diagnosis are raised SIGN guidelines recommends use of a specific surveillance tools such as the Modified Checklist for Autism in Toddlers (MCHAT) to identify children at 18 months old that are at risk of ASD however it cannot be used to rule out a diagnosis [4].
To ensure an accurate diagnosis of ASD a multidisciplinary approach is taken. Pediatricians and Speech and Language therapists are among those involved. The Autism Diagnostic Interview – Revised (ADI–R) is a reliable diagnostic tool and ensures a thorough history is taken.
The child is also clinically observed often with the Autism Diagnostic Observation Schedule (ADOS) as the experience of interacting with a child can elicit the clinical evidence of ASD fitting the diagnostic criteria of ICD–10 or DSM-1V [4].
There is great interest surrounding the etiology of ASD. Studies of monozygotic and dizygotic twins have reported that autism is highly heritable [2]. In four studies reviewing the concordance of ASD among twins it was reported that the median value of concordance for both children receiving a diagnosis of ASD was 76% in monozygotic twins in comparison to 0% in dizygotic twins [5].
Approximately 10% of ASD can be explained by a known genetic syndrome. The remaining 90% are known to have “idiopathic autism”. There is a great interest in identifying susceptibility genes. To date 200 have been identified [6]. However, of the genes that have been identified, none are responsible for a large percentage of cases. It has been proposed that multiple genes (with minor effect) together with environmental influences contribute to the heterogeneous neurobehavioral phenotype that is typical of ASD. Karyotype abnormalities found in ASD are associated with dysmorphic features it would be interesting to investigate whether those with “idiopathic” autism in which susceptibility genes have been identified also have associated dysmorphic features [1].
In the last 5 years Chromosome Array Genomic Hybridisation (array CGH) has made it feasible to not only identifies chromosomal abnormalities but also sub-microscopic chromosomal abnormalities. Sub-microscopic alterations or Copy Number Variants (CNV), which are either inherited or de novo, have been reported in 10- 35 % of ASD cases. The most common ASD related CNV’s are the 15q11q13 duplication, the 7q21 duplication and the 16p11.2 microdelection with its reciprocal microduplication [1]. Christian, et al’s paper on sub microscopic genetic abnormalities in ASD reported that duplications were more likely to be inherited and deletions were more likely to be de novo [7].
Gurrieri et al warns that if we do not incorporate the clinical genetics with a critical evaluation of the autism phenotype then the current research into genetics of autism will be in vain [1].
Aims of study
1. In a well-defined population of children who presented to the multi-disciplinary clinics in the Lothian’s with a diagnosis of ASD we will identify those children who have a chromosome microdeletion or micro-duplication.
2. We will identify specific genes with micro-deletions or micro-duplication and so draw correlations between the genetic abnormality, whether it was de-novo or inherited and the phenotype that the child presented with.
Hypothesis
We predict that sub-microscopic deletions are more likely to be de-novo and therefore present with a phenotypically more severe ASD whereas the sub-microscopic duplications are more likely to be inherited so we predict that they will have a stronger family history and a phenotypically milder disease.
Methods
Study design
Study participants: The following inclusion criteria were used: the patients must be under 18 years old, have a diagnosis of ASD and a genetic diagnosis of either a sub-microscopic deletion or duplication. The children with ASD were diagnosed at a multidisciplinary Communication Clinic in Lothian’s which is run in accordance with the SIGN guidelines on the assessment and diagnosis of ASD [4].
We identified patients by interrogating the Support Needs System (SNS) database for children with a diagnosis of ASD and a chromosomal abnormality. The SNS database is an electronic system that records information about children and young people with additional support needs. Its monitors their progress and ensures access to the appropriate services. It has been implemented in 12 of Scotland’s NHS boards with NHS Lothian having a high level of implementation and utilisation. It is a clinical tool but can also be used for data extraction. A child’s details can only be recorded on the SNS with permission of the parent or guardian.
Community Child Health pediatricians in Edinburgh were also contacted individually to enquire if children who had recently presented to their clinics would fit the inclusion criteria. This identified 89 patients whose notes were assessed according to the inclusion criteria. Of these 6 were excluded as they did not receive a diagnosis of ASD, 43 were excluded because they did not have a genetic diagnosis of a sub-microscopic deletion/duplication and 6 notes did not arrive within the timeframe of data collection. This left 34 patients who were included. One patient had a deletion and duplication so was included under both headings thus giving 35 data sets for analysis.
Data collection
A comprehensive clinical description of each child was recorded in a spreadsheet detailing the child’s phenotype. See Appendix 1.
The antenatal history including whether drugs and alcohol were taken during pregnancy, if the mother experienced depression, infection or bleeding during pregnancy and the parental age at birth were recorded as these have been shown to be pre-natal factors with the strongest evidence for an association with autism risk [8]. The birth history was recorded including whether the child was pre-term (<34 weeks) or term (>34 weeks), the birth weight (kg) and birth trauma were recorded as prematurity, low birth weight and birth trauma have been reported to be autism risk factors [8]. Higher rates of CNV’s have been found in individuals with co- existing structural anomalies and dysmorphisms [9] so these will be documented. The patient’s weight, height and head circumference will be recorded in centiles at the time of diagnosis as it has been reported that macrocephaly (>97th centile) and increased height and weight is found in ASD [10,11]. A history of developmental regression, a diagnosis of a learning disability and epilepsy will also be recorded as these are frequently reported in ASD [4]. A family history of ASD, learning difficulties and a psychotic illness will be recorded as recommended by SIGN guidelines [4]. The child’s sensory behaviors will be recorded as these are included in the general warning signs of ASD [4].
Genetics collection
Blood samples that have previously been obtained from all the children have undergone molecular genetic studies including DNA extraction and array CGH screening for sub-microscopic deletions/ duplications. Some genetic data was missing from the files so the genetics department was contacted to provide the genetic test results of 7/34 children and the parental genetics for 12/34 children.
Results
The 34 patients that were included comprised of 71% males and 29% female. The study included 63% deletions and 37% duplications. Chi-square test of association investigated if there was a statistically significant (p<0.05) association between having a deletion or duplication and the risk factors for ASD that were recorded in the spreadsheet. Odds ratios were also calculated.
Duplications were four times more likely to be inherited and deletions were more likely to be de novo however this was not statistically significant p=0.17 odds ratio= 4.44 95% CI (0.62-32.41) (Figure 1.0).
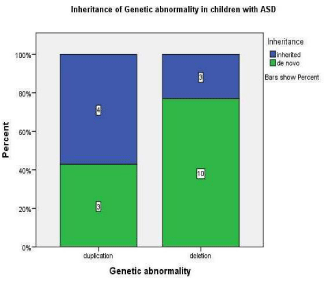
Figure 1: This graph demonstrates that in children with ASD and
microduplication 57% were inherited and 43% were de novo whereas in
children with ASD and micro deletion 23% were inherited and 76% were de
novo
Children with a deletion were six times more likely to have experienced a complications during birth when compared to a duplication p=0.116, odds ratio 6.0095% CI (0.64-55.95) (Figure 1.1). Mothers of children with deletions were nearly four times more likely to bleed during pregnancy or whilst giving birth compared to duplications p=0.37, odds ratio 3.57 95% CI (0.36-35.45) (Figure 1.2). Those children with a deletion were more likely to have a lower birth weight in comparison to duplications p=0.13 (Figure 1.3).
A sub-microscopic deletion in comparison to a duplication doubled the odds of having a structural anomaly p=0.478, odds ratio 2.00 95% CI (0.40-9. 02) (Figure 2.1). Children with deletions were three times as likely to show dysmorphic features p=0.11, odds ratio 3.25 95% CI (0.73-14.40) (Figure 2.2).
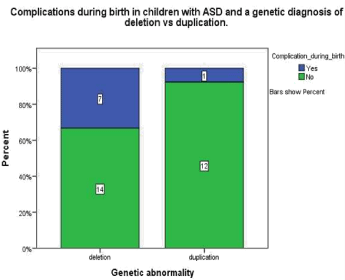
Figure 1.1: This graph demonstrates that in children with ASD and a genetic
diagnosis of submicroscopic deletion 33.3% experienced complications
during birth whereas in children with a diagnosis of ASD a submicroscopic
duplication 7.7% experienced complications during birth.
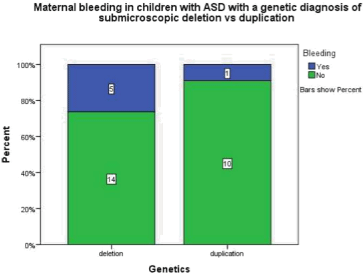
Figure 1.2: This graph demonstrates maternal bleeding either during
pregnancy or labour in children with ASD and a genetic abnormality of
a submicroscopic deletion is 26.3% where as in children with ASD and a
submicroscopic duplication 9.1% had an obsterical haemorrhage.
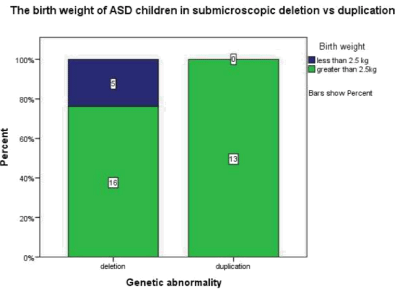
Figure 1.3: This graph demonstrates that in those children with ASD and a
submicroscopic deletion 23.8% had a birth weight less than 2.5kg whereas
the children with duplication all had a birth weight greater than 2.5kg.
Children with a deletion were four times as likely to have a learning disability compared to children with a duplication p=0.16, odds ratio 4.75 95% CI ( 0.719-31.37) (Figure 2.3).
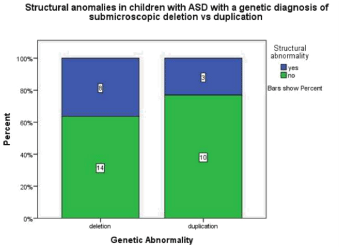
Figure 2.1: This graph demonstrates that children with ASD and a genetic
diagnosis of a submicroscopic deletion 36.4% have a structural anomaly
where in children with ASD and a diagnosis of a submicroscopic duplication
23.1% have a structural anomaly.
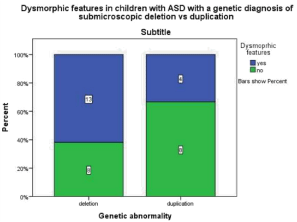
Figure 2.2: This graph demonstrates that in children with ASD and a genetic
diagnosis of a submicroscopic deletion 61.9% were reported to show
dymorphic features whereas in children with ASD and a genetic diagnosis
of a submicroscopic duplication 33.3% were reported to show dysmorphic
features.
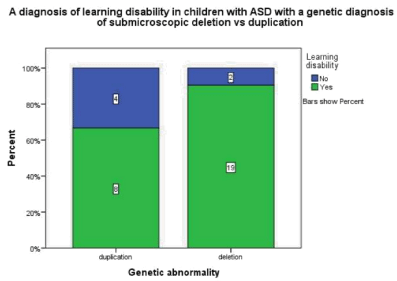
Figure 2.3: This graph demonstrates that in children with ASD and a genetic
diagnosis of a submicroscopic deletion 90.5%also had a diagnosis of a
learning disability whereas in children with ASD and a genetic diagnosis of a
submicroscopic duplication 66.7% also had a learning disability.
Children with a duplication were found to be more than twice as likely to have a family history of ASD p= 0.42, odds ratio 2.5 95% CI (0.53-11.89) (Figure 3.1).
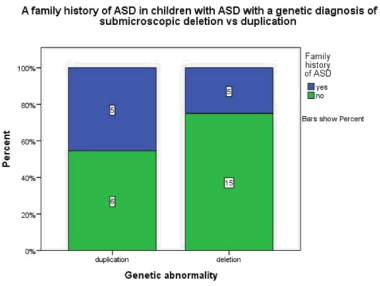
Figure 3.1: This graph demonstrates that in children with ASD and genetic
diagnoses of a submicroscopic duplication 45.5% have a 1st degree relative
family history of ASD whereas in children with ASD and a genetic diagnosis
of a submicroscopic deletion 25% have a1st degree relative family history
of ASD.
The Mann- Whitney U test assessed if deletions were associated with an increased height and weight at diagnosis of autism both of which were found to be off no statistical significance p=0.19 and p=0.64 respectively.
Discussion
Study design
In the last five years, array CGH has been developed that has been reported to identify submicroscopic deletion and duplications in 10-35 % of ASD [1]. This case control study compared the submicroscopic deletion and duplication group retrospectively and sought to determine possible correlations between the genetic abnormality, whether it was inherited or de novo and the child’s phenotype. Case control studies are useful for rare diseases where there is a small cohort of patients as was the case in this study [12]. The study consisted of 71% males and 29% females. This male predominance is widely reported [13].
The data was collected from children’s notes before they had been given a diagnosis of ASD or a genetic diagnosis thus reducing recall bias. The study group was comprised of children on the SNS database that fitted the inclusion criteria and although this is widely utilised in the Lothian’s some children may have been missed as they were not on the SNS database.
A retrospective study is reliant on the quality of the case notes. In this study some patients had missing data as these details had not been recorded in their files. This may be due to some children being born in other countries.
Genetics findings
The primary aim of this study was to report whether the children had deletion or duplications and whether these were inherited or de novo. This study has demonstrated that duplications were more likely to be inherited and deletions were more likely to be de novo (OR= 4.44), however this was not statistically significant. This may be due to the small sample size of 34 and the fact that 12/34 parental genetic details were unknown as some parents had not presented for genetic testing.
Twelve out of the thirty four sub microscopic deletions or duplications in our study have been reported as genetic “hot spots” in the ACMG practical guidelines indicating that these are causal in ASD [14].
We reported that children with a 1st degree family history of ASD were twice as likely to have a duplication compared to a deletion. This adds weight to the fact that duplications are more commonly inherited. Christian, et al reported that in a study of 397 patients with ASD 51 CNV’s, that were not present in the controls, were detected in 46/397 ASD patients (11.6%). Of these 22% were deletions and 78% were duplications. In those with deletions 60% were inherited and 40% were de novo and in those with duplication 86% were inherited and 14% were de novo [7]. This demonstrates that the vast majority of duplications were inherited in their study, however a large proportion of deletions were also inherited despite a much greater proportion of deletions being de novo in comparison to duplications. This may be due to a much smaller number of cases having a deletion thus skewing the results.
Array CGH was only brought into standardized practice five years ago this may explain why Christian et al reported a greater percentage of incidental duplications than deletions. In our study all children under five had the genetic test done routinely after receiving a diagnosis of ASD however the children older than five that have subsequently been tested appear to have a more “severe” autism with potentially more structural anomalies and dysmorphisms. As 86% of the children in the study were over five it is not known if this may have influenced findings. This may explain why the deletion group is larger than the duplication group. It will be interesting to investigate in the future if the CNV’s found on routine testing are more commonly incidental findings.
Association between peri-natal and neonatal risk factors of ASD and genetics
The second aim of this study was to draw correlations between the genetic abnormality and the child’s phenotype.
We reported the odds ratio of having a deletion in comparison to a duplication was increased with complications during birth (OR= 6.00), maternal bleeding (OR= 3.59), structural anomalies (OR= 2.00) and dysmorphisms (OR= 3.25) as predicted. A meta-analysis of 64 studies compared the Perinatal and neonatal factors of ASD compared to a non-autistic control group. They reported positive associations between low birth weight (p=0.002), birth complications (p<0.05) and maternal hemorrhage (p=0.003) and ASD [15]. The increased odds ratio of these children with sub-microscopic deletions having these three risk factors in comparison to children with duplication indicates that their ASD is more likely to be de novo and possibly a result of factors during pregnancy and birth. The results we have reported may well be statistically significant if replicated on a larger scale.
Several studies in Gardener, et al meta-analysis used composite scales to reflect compromised perinatal and neonatal health. It helps solve the problem of low statistical power attributed to rare perinatal and neonatal complications [15]. There is currently insufficient evidence to state that one specific peri-natal or neonatal risk factor is implicated in autism aetiology however the studies using the composite scales suggests exposure to multiple neonatal risk factors may increase risk. To increase the statistical power of our results we could have used a composite scale to give an overall impression that perinatal and neonatal risk factors increase the risk of having a deletion as opposed to duplication.
Phenotype of children with sub microscopic deletion versus duplication
Children with a deletion were more likely to be born with structural anomalies (OR= 2.00) and dysmorphisms (OR= 3.25). Gardener, et al reported a statistically positive association between congenital malformations and ASD (p=0.001) with a high heterogeneity p= 0.87 which may be attributed to different definitions of a congenital anomaly.
In the deletion group 20% had a congenital heart defect whereas none had a congenital heart defect in the duplication group. This strengthens the hypothesis that deletions are more likely to be de novo and therefore have a more “severe” phenotype as they are more commonly sporadic genetic abnormalities.
Children with a deletion were four times more likely to have a diagnosis of a learning disability in addition to ASD than children with a duplication further demonstrating a more severe phenotypical presentation of ASD in those with a deletion.
Head circumference, height and weight in ASD with genetic diagnosis
A meta-analysis of 15 reports explains that the inconsistent findings in the literature between the relationship of head circumference and ASD are due to age-related changes in brain development. They report a consistent pattern of brain size being reduced at birth, dramatically increased within the first year of life and then plateauing so that by adulthood the majority of cases are within the normal range [16]. Our recorded measurements were taken at the time autism was diagnosed and the inconclusive results may reflect the fact that the increased head circumference, height and weight has been reported in the first year of life and plateauing thereafter. Our study was retrospective and we can only work with the data previously recorded however if a similar prospective study was carried out these measurements could be taken throughout the first year of life.
Conclusion
This case control study of 34 children with a diagnosis of ASD and either a sub-microscopic deletion or duplication has shown that although not statistically significant duplications were more likely to be inherited and have a family history of ASD and deletions were more likely to be de novo. Children with a sub-microscopic deletion showed increased neonatal risk factors for ASD.
They were also more like to have a more “severe” phenotype of ASD with an increased prevalence of a learning disability, structural anomalies and dysmorphisms in comparison to children with duplication. Identification of these genetic abnormalities in children may allow early targeted intervention.
Limitations of Study & Further Research
This study had a very small cohort of 34 patients and unfortunately 12/34 parental genetics were unknown. A similar study on a larger scale would further our knowledge of the phenotypical presentation of different genetic abnormalities.
It would be more likely to yield positive statistical results which would add power to the increased odds ratios we have reported in this study.
Further research is required in to the genetics and phenotypical correlations in ASD if we are to provide earlier diagnosis of ASD and intervention which is widely believed to improve outcome.
References
- Gurrieri F. Working up autism: the practical role of medical genetics. American Journal of Medical Genetics. 2012; 160C: 104-110.
- Caronna EB, Milunsky JM, Tager-Flusberg H. Autism spectrum disorders: clinical and research frontiers. Arch Dis Child. 2008; 93: 518-523.
- O’Hare A. Autism spectrum disorder: diagnosis and management. Archives of Disease in Childhood Education & Practice. 2009; 94: 161-168.
- SIGN. Assessment, diagnosis and clinical interventions for children and young people with autism spectrum disorders. 2007.
- Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 2011; 156B: 255-274.
- Heil KM, Schaaf CP. The genetics of Autism Spectrum Disorders--a guide for clinicians. Curr Psychiatry Rep. 2013; 15: 334.
- Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, Karamohamed S, et al. Novel Submicroscopic Chromosomal Abnormalities Detected in Autism Spectrum Disorder. Biol Psychiatry. 2008; 63: 1111-1117.
- Gardener H, Spiegelman D, Buka SL. Prenatal risk factors for autism: comprehensive meta-analysis. The British Journal of Psychiatry. 2009; 195: 7-14.
- Schaefer GB, Mendelsohn NJ, Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genetics in Medicine. 2008; 10: 301-305.
- Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G, et al. Head circumference and height in autism: A study by the collaborative program of excellence in autism. American Journal of Medical Genetics Part A. 2006; 140A: 2257-2274.
- Chawarska K, Campbell D, Chen L, Shic F, Klin A, Chang J. Early generalized overgrowth in boys with autism. Arch Gen Psychiatry. 2011; 68: 1021-1031.
- Mann C. Observational research methods. Research design II: cohort, cross sectional and case-control studies. Emergency Medicine Journal. 2003; 20: 54-60.
- Brugha T, McManus S, Meltzer H, Smith J, Scott F, Purdon S, et al. Autism spectrum disorders in adults living in households throughout England: Report from the adult psychiatric morbidity survey 2007. The Health & Social Care Information Centre, Social Care Statistics. 2009.
- Schaefer GB, Mendelsohn NJ, Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genetics in Medicine. 2013.
- Gardener H, Spiegelman D, Buka SL. Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics. 2011; 128: 344-355.
- Redcay E, Courchesne E. When is the Brain Enlarged in Autism? A Meta- Analysis of All Brain Size Reports. Biol Psychiatry. 2005; 58: 1-9.