
Review Article
J Bacteriol Mycol. 2021; 8(2): 1165.
Candida-Associated Gastric Ulcer Revisited
Sasaki K*
Department of Internal Medicine, Midtown Medicare Clinic, Japan
*Corresponding author: Sasaki K, Department of Internal Medicine, Midtown Medicare Clinic, 76-1, Nakada, Kanetani, Matsuyama, Ohsaki, Miyagi, 987- 1303, Japan; Tel: +81-80-1817-8414; Fax: +81-229-55- 4673; Email: kydosarnymai@aria.ocn.ne.jp
Received: January 29, 2021; Accepted: February 19, 2021 Published: February 26, 2021
Abstract
Candida-associated gastric ulcer occurs not only in debilitated but healthy individuals. Though had been reported to demonstrate nothing but nonspecific endoscopic features, it occasionally exhibits a typical finding I designated a candidarium. The natural history of the disease had not been clarified and the recurrence had not been described. However, I demonstrated that the ulcer not only occurs but also recurs in a different site with a different shape in a non-diabetic, H. pylori-negative patient, who has not taken NSAIDs, antibiotics, antineoplastic agents, or systemic corticosteroids, advocating that, contrary to the prevailing opinion, Candida is no innocuous bystander but an etiologic perpetrator. It has recently been shown to secrete a cytolytic PFT, candidalysin, into a pocket in the epithelium after penetrating into it to activate MAPK/MKP1/ c-Fos pathway, triggering release of damage as well as immune cytokines in OPC and VVC. While candidalysin, exerting an effect even on the adjacent oropharyngeal cells, directly injures the tissue with damage cytokines, immune counterparts activate PMNs to eventually terminate inflammation. Though the epithelial response to the fungus is different from organ to organ, it invades into and induces necrotic cellular damage to the IECs through the toxin to translocate: the action of candidalysin is proven not only on the squamous but on the columnar epithelium. Since, by analogy with intestinal candidiasis, it is never difficult to speculate that the PFT inflicts such damage to the gastric mucosa, a theoretically strong possibility has come up that Candida-associated gastric ulcer is actually Candida-induced.
Keywords: Candidarium; Candida-Associated Gastric Ulcer; Candida- Induced Gastric Ulcer; Candidalysin; H. Pylori-Negative Gastric Ulcer; Recurrent Gastric Ulcer
Abbreviations
H. pylori: Helicobacter Pylori; NSAID: Non-Steroidal Anti- Inflammatory Drug; C: Candida; PFT: Pore-Forming Toxin; MAPK: Mitogen-Activated Protein Kinase; MKP1: MAPK Phosphatase 1; VVC: Vulvovaginal Candidiasis; GI: Gastrointestinal; SMT: Submucosal Tumor; PPI: Proton Pump Inhibitor; RHOE: Reconstituted Human Oral Epithelium; Sap: Secreted Aspartyl Proteinase; JNK: c-Jun N-Terminal Kinase; ERK: Extracellular Signal- Regulated Protein Kinase; NF-κB: Nuclear Factor Kappa-Light- Chain-Enhancer of Activated B Cells; PMN: Polymorphonuclear Leukocyte; EGFR: Epidermal Growth Factor Receptor; MMP: Matrix Metalloproteinase; PAMP: Pathogen-Associated Molecular Pattern; DAMP: Danger-Associated Molecular Pattern; OPC: Oropharyngeal Candidiasis; IEC: Intestinal Epithelial Cell; PRR: Pattern Recognition Receptor; TLR: Toll-Like Receptor; NKT: Natural Killer T Cell; ILC3: Type 3 Innate Lymphoid Cell; nTh 17: TCRβ+ “natural” Th 17 Cell
Introduction
Though a commensal colonizer ubiquitously detected in the normal human oropharynx to GI tract throughout [1], Candida causes infections in the tract under certain conditions, which are more widespread than previously recognized [2,3]. The stomach is the second most frequently involved organ following the esophagus [2]. Gastric candidiasis is classified into thrush, nodular, and ulcerated type [4]. Though usually seen in immunocompromized hosts [2,3], the third type, Candida-associated gastric ulcer, also occurs in apparently healthy individuals [5,9] with very widely different frequency contingent on the researchers [8-12]. The natural history of the disease had not been clarified and the clinical significance of the fungus remains to be elucidated [2] so that the treatment has not yet been established. It had been reported to be no longer detected once the ulcers were healed and no recurrence of the disease had been described [9,11]. I [13], however, reported a case of the disease relapsing in a different site with a different shape in an H. pylorinegative, non-diabetic patient with no antecedent peptic ulcers or history of the lesions, who had not taken NSAIDs, antibiotics, antineoplastic agents, or systemic corticosteroids, speculating that it plays an etiologic role.
Given the recently clarified mechanism of pathogenicity of candidiasis in the oropharyngeal, vulvovaginal, and intestinal fields, I have drastically updated my former review of Candida-associated gastric ulcer to evaluate the validity of the speculation in the light of the recent microbiological, molecular biological and immunological findings.
Candida Infection of the GI Tract
Candida is totally lacking a lifestyle outside the human body and establishes a colony in the human oropharynx and in the GI and vaginal tracts. Langenbeck [14] is credited with having discovered it in the cadaveric intestine in 1839 and now C. albicans is proven to be the most frequent symbiont fungus in the oropharynx to the GI tract of the normal human adults [1]. It increases both in frequency and concentration directed toward the anus: it was detected in 27% of the oropharynx samples obtained by swabbing, 43% of the jejunal and 50% of the ileal aspirates, and 59% of the fecal specimens [1].
No fungal growth, whether it is Candida or not, was detected in the gastric juice with pH value lower than 4.0 and the positive rate of fungal recovery was significantly increased with the elevation of the acidity of the juice [15]. Colonization of Candida in the stomach was observed more frequently in older patients [11] and in patients with hypoacidity [16]. It is also demonstrated to be the most common fungus causing significant, histologically proven GI infection in the debilitated patients: the infection was detected in 13% of the patients with myeloproliferative diseases and in 1.5% of those with solid tumors, respectively [2]. It involved all segments of the GI tract but the most frequently afflicted organ was the esophagus followed by the stomach [2].
Candida infection is more widespread than previously recognized [2,3], shown to occur not only in debilitated but also apparently healthy individuals [3]. Gastric candidiasis is shown to affect 0.96% of all patients undergoing endoscopy and to be more common in men and the elderly [4]. Minoli et al. [4] endoscopically classified the disease into 3 forms: thrush, nodular, and ulcerated, each accounting for 42%, 31%, and 27%, respectively. The first type presents itself as a white or green-white, readily removable membrane of variable extension spreading over the inflamed mucosa in various locations. The second is described as nodular projections of a few milliliters in diameter overlaid by the remarkably inflamed mucosa, mainly located in the antrum. The third has no particular endoscopic features distinct from other peptic ulcers.
Candida-Associated Gastric Ulcer Until Yesterday
Candida-associated gastric ulcer is regarded as the above mentioned ulcerated-typed gastric candidiasis, the diagnostic criterion of which is demonstration of infiltration of the tissue or ulcer slough by the hyphae [5,8,10]. It is also seen in apparently healthy individuals [5-9] with quite widely different frequency according to the researchers [8-12] ranging from 0.12% to 36% of gastric ulcer. The species in the genus reported to be associated with gastric ulcer are albicans, tropicalis, parapsilosis, krusei, and pseudotropicalis in decreasing order of frequency [10]. The natural history of the disease had remained to be clarified [2] and the disease has been reported to engender no specific symptoms [8]. As Minoli et al. [4] described, the endoscopic features of the disease had been asserted to be nonspecific [9,11].
Whereas some cases of the ulcer have been reported to have spontaneously healed [12], it has been reported to have low healing rate [6,11]. The rate of decrease in ulcer diameter, an indication of lesion healing, was slower in patients whose stomachs were significantly colonized by the fungus as compared with patients who were not [17]. Brozozowski et al. [18] demonstrated that persistent colonization with it in the stomach of rats suffering from ulcer induced by acetic acid, which were inoculated with the fungus, was achieved in those treated with antisecretory agents or NSAIDS and that such candidiasis reduces gastric acid secretion, while delaying ulcer healing possibly due to the impairment in the gastric blood flow in the ulcerated area and enhanced expression and release of IL-1β and TNFa. Reviewing Crohn’s disease, ulcerative colitis, and gastric and duodenal ulcers, Kumamoto [19] points out that significant colonization with it is associated with more severe disease and the colonization delays healing of inflammatory lesions and inflammation promotes colonization, producing a vicious circle.
The fungus had been reported to be no longer detected once the ulcers were healed even without antifungal treatment and no recurrence had been described [9,11]. Though the clinical significance of it has not yet been elucidated, ---- so that the appellation of the disease is modified by an ambiguous adjectival past participle “associated”---- it was not regarded as directly etiologic in development of ulcer but was considered to possibly aggravate or perpetuate ulceration [5,6,8].
I designate such a state of the recognition of the disease Candidaassociated gastric ulcer until yesterday after one of Dr Jared Diamond’s masterpieces. The proofs that it exacerbates gastric ulcer or delays its healing are considered to be the proofs that it has the ability to damage the gastric tissues but they by no means deny that it initiates peptic ulcers. No direct evidences, however, have been obtained so far that it is ulcerogenic.
Candida-Associated Gastric Ulcer Today
I [13], however, presented a then unreported case of Candidaassociated gastric ulcer relapsing in a different site with a different appearance in a non-diabetic, H. pylori-negative patient with no antecedent peptic ulcers or history of the lesions who had not taken NSAIDs, antibiotics, antineoplastic agents or systemic corticosteroids. The first ulcer was detected in an 87-year-old, Japanese housewife complaining of anorexia as a medium-sized, SMT-like elevation overlaid with the erythematous mucosa with an oval, deep, central ulceration covered with thick, whitish exudate I designated a candidarium [20] (vide infra) on the greater curvature of the upper gastric body (Figure 1). No signs of candidiasis were detected in the oropharynx through esophagus or in the duodenal bulb through descending part of the duodenum. She had endoscopically been shown to have no such lesions or scars in the upper or no signs of candidiasis in the lower GI tract 2 months before. She had no past history of peptic ulcers. Biopsy showed no malignancy or H. pylori but countless hyphae in the ulcer slough (Figure 2), which were proven to be C. tropicalis by culture. She, though diagnosed as having Candida-associated gastric ulcer, refused to be treated.
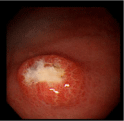
Figure 1: Endoscopic photograph showing a medium-sized, SMT-like
elevation with an oval, deep, central ulceration covered with thick, whitish
exudates designated a candidarium (see text) on the greater curvature of the
upper gastric body.
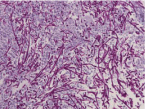
Figure 2: Light micrograph of the biopsied specimen from the candidarium
in Figure 1 demonsrating innumerable hyphae in the ulcer slough (PAS/
diastase, original magnification x400).
The lesion was shown to have spontaneously turned into a white scar (Figure 3) 10 months later, when she complained of heartburn, but another large, oval, deep ulcer was detected covered with thick, whitish exudate surrounded by the markedly swollen margin on the lesser curvature of the lower gastric body (Figure 4). Biopsy demonstrated numerous hyphae again (Figure 5). H. pylori was not detected by histologic examination or rapid urease test. No findings of candidiasis were recognized in the oropharynx through other parts of the upper GI tract. She was diagnosed with Candida-associated gastric ulcer recurrent in a different location with a different appearance. The lesion was proven to have turned into a red scar (Figure 6) in 6 weeks with administration of a PPI without an antifungal agent and into a white in 3 months (Figure 7) [13].
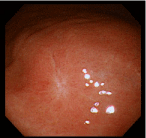
Figure 3: Endoscopic photograph showing the white scar of the original ulcer.
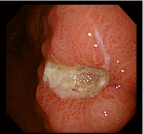
Figure 4: Endoscopic photograph showing a recurrent ulcer on the lesser
curvature of the lower gastric body.

Figure 5: Light micrograph of the biopsied specimen from the recurrent
ulcer demonstrating numerous hyphae of Candida in the ulcer edge. (PAS/
diastase, original magnification x400).
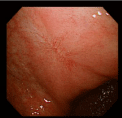
Figure 6: Endoscopic photograph showing the red scar of the recurrent ulcer.
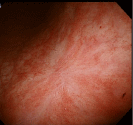
Figure 7: Endoscopic photograph showing transition from the red to white
scar in 3 months.
While endoscopic features of Candida-associated gastric ulcer had been asserted to be nonspecific [4,9,11], an SMT-like lesion with a deep, central ulceration has been regarded as specific by some Japanese authors [21,22]. In my case, the original lesion did indeed assume such a form [13] (candidarium) [20]. Though might be peculiar to the race because such an appearance has been detected only in Japanese patients [21,22], so far, it is considered suggestive of, or even possibly pathognomonic for, the gastric ulcer. I [20] propose to designate the lesion a candidarium (Candida hill; Candida, the genus of the fungus, originally a Latin feminine adjective meaning bright + arium, a Latin neutral substantive-forming suffix usually denoting a place) after a formicarium (ant hill; formica, a Latin feminine noun signifying an ant).
Whereas the disease has been reported to have low healing rate [6,11], some cases of the disease have been reported to have spontaneously healed [12]. In my case, the original lesion was found to have spontaneously healed and the recurrent one was in no way intractable [13]. The intractability of the disease may be affected by other factors than the fungus per se, such as H. pylori, NSAIDs, or specifically impaired immune response of a host to the fungus because such factors were not inspected in the cases reported to be intractable [6,11].
The fungus had been reported no longer detected once the ulcers were healed [9,11] and the natural history of Candida-associated gastric ulcer had still been to be elucidated [2]. My case [13], however, is the first in the world, which was demonstrated to have recurred not only in a different site but with a different appearance and followed up from before development of the original ulcer till complete cicatrization of the recurrent, disclosing the natural history of the disease: the fungus is not considered an innocent bystander, which is secondarily detected in such an already existing ulcer, as the lesion induced by acetic acid [18].
Candida-Associated Gastric Ulcer from Tomorrow
Candida-associated gastric ulcer is considered Candidainduced
Detected both in the original and recurrent lesions in a nondiabetic, H. pylori-negative patient with no antecedent ulcers who had not taken NSAIDs, antibiotics, antineoplatic agents, or systemic corticosteroids, Candida was considered, contrary to the prevailing opinion, to play an etiologic role in ulcer formation [13]. I speculated that the fungus exerted the direct gastric mucosal damage in the setting of the compromized mucosal defense in the elderly patient. Such a tissue injury could be aggravated by the presence of gastric acid, however weakened by old age. Zelante et al. [23] demonstrated that intragastric inoculation of C. albicans engenders epithelial necrosis through activation of IL-23/IL-17 pathway in mice. Such a situation is considered to result in ulcer, in the slough of which numerous hyphae are detected.
I [20] infer as follows. This is not simply only one case of Candida-associated gastric ulcer but is, as it were, the only one fossil luckily unearthed in its integrity, so far. Though dug out, other ones are marred by H. pylori, NSAIDs, antibiotics, antineoplastic agents, or systemic corticosteroids in the extremely specific environment, the stomach, in which peptic ulcer occurs not rarely from different causes, so that they leave so much to be desired to reflect the whole picture of the bona fide Candida-associated gastric ulcer. This is not a peculiar one or exception, which is incidentally discovered by a whimsical endoscopist but the theoretically predictable missing link, which connects Candida-associated gastric ulcer in the Dark Age to Candida-induced gastric ulcer in the era enlightened by the splendid future researchers. It is an ideal model, which represents all the natural gastric ulcers associated with Candida infection and is never excavated frequently.
It is the ability of the fungus to switch from the yeast to hyphal form, by which the different genes of it are expressed, that is crucial in causing pathogenicity at the mucosal surfaces [24]. Wachtler et al. [25] demonstrated that C. albicans hyphae can invade TR146 epithelial cells through two distinct mechanisms, induced endocytosis and active penetration, and that the latter is the dominant invasion route. Jacobson et al. [24] showed that it is only the active penetration of the hyphae, which causes epithelial damage. Silva et al. [26] demonstrated that C. tropicalis hyphae are highly invasive with the ability to induce tissue damage in a RHOE system with no involvement of Saps, the key hydrolytic enzymes secreted by the fungus, implying the existence of a still unknown mechanism.
Moyes et al. [27] demonstrated that oral epithelial cells, discriminating between the yeast and hyphal forms of C. albicans, exhibited a biphasic MAPK response. The first phase of the response is provoked when fungal burdens are light, which constitutes weak activation of all MAPK pathways, p38, JNK, and ERK1/2 together with NF-κB independent of the fungus morphology. This results in activation of the transcription factors NF-κB and c-Jun through ERK1/2 and JNK, which is not strong enough to induce immune activation, allowing the fungus to remain in the commensal state. The second phase is observed when fungal burdens are heavy, which is dependent upon hypha formation, constituting activation of the transcription factor c-Fos through p38. In addition, the hyphae activate MKP1 through ERK1/2 pathway, which acts to regulate p38 and JNK signaling. MAPK/MKP1/c-Fos activation results in immune activation and secretion of proinflammatory cytokines. Such events ultimately lead to PMN recruitment and fungal clearance or burden reduction, namely a return to the commensal state [28].
Moyes et al. [29] have recently succeeded in clarifying how C. albicans induces epithelial inflammatory responses and cell damage during mucosal infections: they identified and characterized the first cytolytic, a-helixed, peptide PFT isolated from any human fungal pathogen as the hyphal factor critical for epithelial immune activation and mucosal infection of the fungus. The hyphae invade the epithelium to create invasion pockets, into which they secrete the toxin Ece1-III, the third segment of Ece1 designated “candidalysin” by Moyes et al. [29], thereby triggering the above mentioned second phase of MAPK response, which issues in production of immune regulatory cytokines, such as IL-6, G-CSF, and GM-CSF, at the sublytic concentrations. As the toxin increases in amount to reach the lytic concentration, it damages the epithelium to release damage cytokines, such as IL-1a. Candidalysin, if produced sufficiently, appears to exert an effect on the epithelial surface outside of the invasion pocket as well as on the adjacent cells not in contact with the hyphae [29].
It resides as a discrete cryptic sequence within a larger 271- amino-acid parental preproprotein, Ece 1p encoded by C. albicans ECE1 gene. Kexin-like proteinases, but not Saps, initiate a two-step posttranslational processing of the preproprotein to produce the PFT. Endoproteinase Kex 2p-mediated proteolysis of Ece 1p at Arg61 and Arg93 is required to generate immature toxin from it, followed by carboxypeptidase Kex 1p-mediated removal of carboxyl arginine residue to generate mature one. Kex 2p is a member of a family of eukaryotic proprotein proteases possessing catalytic domains homologous to those of the bacterial counterpart subtilisin family. These subtilisin/kexin-like proteases have been implicated in the activation of various bacterial toxins [30].
EGFR is identified as a critical component of candidalysintriggered immune responses: inhibition of EGFR impairs the PFTinduced MAPK signaling and release of PMN-activating chemokines and diminishes PMN recruitment. Calcium influx through invasion pockets provokes MMP activation and expression, which leads to cleavage of EGFR ligands, predominantly epiregulin and epigen. These events result in EGFR activation inducing MAPK signaling. Thus, a PAMP-independent mechanism of immune stimulation is identified [31].
It is demonstrated that not only oral but vaginal epithelial cells can differentially sense and respond to yeast and hyphal forms of C. albicans with modestly different signaling mechanisms, cytokine secretion, and hyphal activation thresholds [32,33]. Roselletti et al. [34] demonstrated that the intracytoplasmic activation of NLRP3 inflammasome complex plays a critical pathogenic role in human vulvovaginal disease engendered by C. albicans but they also reported that, though the hyphae account for a relatively minor component of the fungal forms in the vagina, ECE1 is overexpressed in the organ. Treatment of vaginal epithelial cells with candidalysin induced dose-dependent proinflammatory cytokine responses, damage, and activation of MAPK/MKP1/c-Fos system. Whereas no difference in colonization was shown in mice intravaginally challenged with candidalysin-negative C. albicans strains, significant decrease was observed in PMN recruitment, damage, and proinflammatory cytokine expression compared with controls inoculated with the wild-type fungus. Richardson et al. [35] think that it is possible that the PFT serves as a fungal DAMP capable of inflammasome activation. Thus, candidalysin is shown to play a critical role in inducing immunopathological signaling not only at the oral but also at the vaginal mucosa.
Thus, the possibility has been augmented that C. albicans induces toxic action in mucosal infections just as bacteria do. A logically definitive possibility has opened up that Candida-associated gastric ulcer is provoked by the direct action of the fungus so that the disease should be designated Candida-induced ulcer instead of Candidaassociated.
The PFT-MAPK/MKP1/c-Fos system is considered to work in not only C. albicans-but Non- C. albicans Candida (NCAC)-associated gastric ulcer
Candidalysin-MAPK/MKP1/c-Fos system was established only as regards C. albicans OPC in the TR146 human buccal epithelial squamous cell carcinoma line system in vitro and with the use of female Balb/c mice OPC model modified for investigation of early infection events in vivo [29] and in terms of vulvovaginal C. albicans infection in the A431 human vulvar epidermoid carcinoma cell line system in vitro [32] and in the C57BL/6 mouse system in vivo [35], respectively. Does what comes to pass in the squamous epithelium also occur in the gastric mucosa?
Lee et al. [36] demonstrated that C. albicans pre-vacuolar protein sorting gene VPS4 is required for extracellular secretion of Sap2p and Saps4-6p and that the C. albicans vps4D null mutant is markedly hypovirulent in a standard murine tail vein model of disseminated candidiasis [37]. Thereafter their group [38] explored the role of the pre-vacuolar secretion pathway mediated by VPS4 in the pathogenesis of epithelial and mucosal infection using a wide range of virulence models, obtaining the results which suggest that VPS4 contributes to several key aspects of oral epithelial but not uroepithelial infection and no major part in the pathogenesis of VVC in contrast to disseminated candidiasis. They [38] concluded that C. albicans VPS4 contributes to virulence according to the specific tissue that is infected.
Vautier et al. [39] performed the following experiments in order to examine which morphology of C. albicans best fits to colonize the organs of the GI tract by analyzing respective fungal burdens at various organs. Mice, which were pretreated with oral administration of antibiotics to reduce the commensal bacterial and fungal microbiota, were perorally infected with wild-type strains (SC5314 and CAI4) as well as strains carrying mutations locking them into the yeast (efg1D/cph1D), or hyphal forms (nrg1D). Animals infected with the hyphal-locked strain showed lower burdens in the small intestine, cecum, and large intestine, whereas those infected with the yeast-locked strains had similar or higher burdens as compared with those infected with the wild types. Those infected with hyphallocked strain of MBY38 demonstrated significantly lower tissue fungal burdens, as well. Colonization in the murine intestinal tract was, therefore, shown to favor the yeast morphology. Whereas in the stomach, both the strains locked into the yeast and into the hyphal form were recovered at lower levels as compared with the wild-type strains. These results suggest that it is essential that the fungus be provided with the ability to shuttle between both the morphologies in order to colonize in this specific organ. Correlated with their reduced fungal burden in the stomach, decreased levels of cytokines, IL-1β, IL-6, and IL-17, which are involved in mediating Th17 responses [40], were detected in the organ of the mice infected with both the yeast- and hyphal-locked strains. Such phenomena were not observed in any other organs [39]. These results endorse preferential infection of the stomach in the experimental model [41] and the fact that the colonization is restricted to the lumen elsewhere in the GI tract [39]. Though Th17 immunity is required for controlling C. albicans infections at the mucosa, the significance of decreased level of these cytokines produced by both the yeast- and hyphal-locked strains colonizing in the stomach remains to be elucidated. No experiments were conducted concerning the possibility that interfering with Th17 responses would influence colonization of the fungus restricted to the organ by blocking the effects of IL-1β or IL-17.
Though, reflecting the organ-specificity in C. albicans infection in the experimental animal models, the predominant morphology of wild-type Candida, which has the ability to shuttle from yeast to hyphal form, differs from infected organ to organ [18], the fungus shows hyphal form in Candida-associated gastric ulcer [4- 6,8,10,12,13,21]. If the fungus can transgress the mucus barrier of the stomach and overcome the inhibitory action of the serum [21] and if candidalysin is not inactivated by gastric acid, at least in an invasion pocket, it is thought to be able to penetrate actively into the gastric mucosa to provoke ulcer by the PFT and damage cytokines produced by MAPK/MKP1/c-Fos pathway [29] with the aid of gastric acid. Though such a phenomenon has not been confirmed in human C. albicans-associated gastric ulcer, it does not appear irrational to infer so.
Adhesion to, invasion into, and damage to the epithelial cells by C. albicans depend not only on fungal morphology and activity but on the epithelial cell type and differentiation stage. Epithelial cells differ in their susceptibility to the fungus [42]. The mucosal surface of the oral and vaginal lumen is not the same as that of the GI tract. The former is made up of the stratified squamous epithelium with the outermost terminally differentiated layer, whereas the latter comprises the single layer of the columnar cells covered with mucus of which mucin is the main constituent.
Not only the mucin Muc5AC which is expressed in the stomach and in the lung but Muc2 from the intestinal mucus and Muc5B from the saliva exert suppressive effect on C. albicans physiology, which suggests that the ability of mucins to manage fungal virulence may be general mechanism present on all mucosal surfaces as part of the innate mucosal immune system [43]. However, the fungal hyphae secrete Sap 2p which degrades mucins [44] and renders them possible to make access to the epithelial surface.
Als3, hypha-specific C. albicans invasin, is reported to play a key role in multiple processes including adherence to and invasion into host cells [45,46]. It is shown to be a key invasin necessary for internalization of the fungus into the monolayers of TR146 [45] and HeLa [24] cells. It facilitates internalization of the fungus through both active penetration and induced endocytosis by the differentiated IECs displaying altered tight junction [45].
Infection of IECs by C. albicans provokes the barrier breakdown related to considerable reduction of the level of the tight junction proteins and their disappearance from the cell-to-cell borders. Host cell lysis is observed at much later time points so that invasion of the fungus and cell damage caused by it are considered separate postinfectional events [47] just as, though adhesion to and subsequent invasion into the oral epithelial cells result in fungal recognition and signal pathway activation, the steps do not translate into epithelial damage and innate immune activation [27,48]. After invasion of the fungus into the IECs, upregulation of the genes is observed involved in PRR downstream signaling, cellular stress, inflammation, viz. MAPK, TNF, and NF-κB signaling pathway [47]. It is candidalysin that induces necrotic cellular damage but not apototic death of the IEC, as it causes damage to the oral [29] and vaginal [24] mucosa, and subsequent translocation of the fungus [49]. The action of candidalysin is proven not only on the stratified squamous mucosa but on the single layer of the columnar epithelium. A combination of hypha formation and candidalysin secretion is required for optimal damage induction of the IECs [49]. Though there exist strong differences between the intestinal and oral epithelial cells in response to C. albicans infection, several genes of the inflammatory response and PRR downstream signaling are similarly activated in both, such as similar time-shifted differential expression of Jun and Fos [47].
By analogy with intestinal candidiasis, interaction of C. albicans with the gastric epithelium may be understood. It is highly probable that the gastric epithelium is reduced to necrosis by candidalysin secreted by the invading fungus with subsequent activation of MAPK/MKP1/c-Fos system together with the aid of gastric acid, which results in Candida-induced gastric ulcer. Though the ways are different how the fungus activates signaling mechanisms from organ to organ, the system is common among them examined so far, which enables epithelium to orchestrate innate immune responses specifically against the fungal hyphae [32].
But does such an event occur only in a case of C. albicansassociated gastric ulcer and not in a case of NCAC? Moyes et al. [33] demonstrated that no NCACs exhibit true hyphal forms but C. dublinensis in vitro. Though Silva et al. [26] reported that C. tropicalis was highly invasive with the ability to induce significant tissue damage, exhibiting the hypha formation, in the same in vitro system, as Moyes et al. [33] used, the latter [33] claimed that invasion and cytokine production by C. tropicalis was significantly lower than that induced by C. albicans in their experimental system and that, although C. tropicalis may form “hyphal-like” structures in vivo to activate epithelial cells, it is unlikely that they will parallel the true hyphae produced by C. albicans et dubliniensis and are thus unlikely to possess the same hyphal moiety shared by C. albicans to activate the epithelial cells via MAPK/MKP1/c-Fos pathway.
I [13] presented a case of C. tropicalis-associated gastric ulcer, in whose slough innumerable obvious hyphae were detected, as in a case of C. albicans-associated gastric ulcer [12]. The similar phenomenon provoked by candidalysin is considered to be generated not only in C. albicans- but also in NCAC-associated gastric ulcer. Since the PFT-MAPK/MKP1/c-Fos system, though there exist various PFTs, is generally recognized in a wide variety of bacterial infections [50], it is unlikely that C. albicans is the sole species which possesses PFT in the genus. It is true that dramatic interspecies variability has been reported to exist in it, so far: C. glablata is asserted to be unable to form hyphae, to have no candidalysin ortholog, and to exhibit no virtual immunopathogenicity in murine model of VVC, yet it clearly provokes the disease in women [51]. It is not at all difficult to deduce that fastidiousness of NCACs renders the obvious hypha formation irreproducible in vitro and that, therefore, the true in vivo events engendered by the fastidious fungi have by no means been able to be investigated in vitro until now. Establishment is expected of the proper in vitro experimental system of NCACs and of in vitro and in vivo “Candida-associated gastric ulcer”.
Immunity against Candida
Differences are reported in immune response requirements at different mucosal sites. Though Hernandez-Santos et al. [52] demonstrated that Th17 cells mount a robust and stable antigen specific adaptive immunity against OPC in mice, Richardson and Moyes [53] stated that the establishment of bona fide immunological memory remains to be fully demonstrated.
On the other hand, the recent studies have challenged the dogma that innate immunity has no memory. Long-term changes of cells of the innate immune system have recently been shown to be provoked by encounters with PAMPs. C. albicans adaptively evolved in the murine GI tract induced the phenotype which not only lost their virulence but protected their host from systemic infections. The protection, developed very early, was not dependent on adaptive immunity but on increased innate cytokine responses, reminiscent of “trained immunity” [54]. Infection of mice with a poorly virulent strain of C. albicans incapable of yeast-hyphal conversion conferred T cell-independent but macrophage-mediated protection against subsequent challenge with a highly virulent strain [55,56]. In addition to the above findings, a sublethal C. albicans infection afforded protection against a subsequent lethal reinfection in a T/B cell-independent, monocyte-dependent manner through functional reprogramming of monocyte via Dectin-1/Raf-1 pathway associated with stable and genome-wide changes in histone methylation [57- 59]. In such trained monocytes, a metabolic switch was demonstrated to aerobic glycolysis, which was crucial for maintenance of trained immunity, as seen in the Warburg effect in the neoplastic cells [60]. Short pre-stimulation of RHOE with heat-killed C. albicans yeast showed a significant increase in the expression of human β-defensins 2 and 3 associated with impaired growth and viability of the fungus [61]. Further clarification of these phenomena is expected.
While about 75% of immune competent women will be afflicted with VVC, almost 9% of them suffer from the recurrent infection [51]. Though Th17 cells are regarded as the predominant cell type that confer protection against OPC, IL-22 and IL-17 do not contribute to immune protection in VVC [62]. It was demonstrated that human oral and vaginal epithelial cells perform different host response to C. albicans: the former by secreting cytokines and chemokines, while the latter by direct fungal killing. But both cells play only a minor role in adaptive immunity against the fungus [63].
Th17 cells secrete interleukins IL-17A and IL-17F, which stimulate a variety of cells to produce antimicrobial peptides and chemokines promoting PMN recruitment and activation [64]. Patients with defects affecting segments of innate and acquired immunity which disrupt the Th17 pathway are reported to develop chronic mucocutaneous candidiasis because they are unable to clear superficial Candida infections [65]. von Bernuth et al. [66] stated that, though humans with inborn MYD88 or IRAK-4 deficiency suffer from a few naturally occurring life-threatening bacterial infections, TLR- and IL-1R-dependent immunity mediated by such genes is crucial only in infancy and early childhood in the natural setting in contrast to the murine counterpart. But Vogelaar et al. [67] reported a case of a young adult with recurrent candidiasis, who had a germline homozygous missense variant in MYD88 and exhibited a defective production of IL-17 upon stimulation with C. albicans.
IL-17 is produced not only by Th17 cells of the classical immune response but also by a variety of innate immune cells, such as γδ-T cells, NKTs, ILC3s, and nTh17s [68]. The immune response of the latter, which is shown to be activated in response to candidalysin secreted by the invading C. albicans hyphae in OPC [69], does not require CARD signaling to be driven unlike that of the former [70]. Though IL-1 is identified as a central player in inducing innate type- 17 immune responses to clear C. albicans infections, lack of IL-1 signaling does not result in complete loss of immunity. As with IL- 1a/β, induction of epithelial IL-36 and its gene expression depend on candidalysin-MAPK/MKP1/c-Fos system. OPC in IL-36-/- mice shows increased fungal burdens and reduced expression in IL-23 gene but not in IL-17 and IL-17-driven genes. Thus, IL-1 and IL-36 represent parallel epithelial cell-driven pathways in immunity to OPC [71].
While ILC3s have been suggested to play a major role in protection against OPC [72], oral-resident nTh17 and γδ-T cells are shown to be the predominant source of IL-17 [73]. On the other hand, pathogenicity of IL-17 and IL-23 was reported in a murine model of gastric candidiasis [23]. Ulcerogenic as the fungus may be, the role of the cytokines should be reevaluated in the light of candidalysin- MAPK/MKP1/c-Fos system because the study was executed before the discovery of the PFT. Immune responses against Candida is species- and organ-specific and effective immunities are so uniquely compartmentalized that the results obtained in the one species or organ will not be necessarily applied to the other.
Conclusion
Owing to the recent advances in microbiology, molecular biology, and immunology, a logically stronger definitive possibility has emerged that the so-called Candida-associated gastric ulcer is Candida-induced. The disease has come to a stage, in which the etiology should be reinvestigated and the disease itself should be reconsidered in the light of not only pathogens’ character but also hosts’ immunological status.
Acknowledgements
I wish to express my gratitude to two distinguished pathologists, Dr Masuda at Miyagi Cancer Society and Dr Iwama at Tohoku Rosai Hospital for their very helpful instructions. This review is dedicated to Prof Andrzej Tarnawski at University of California, who gently acclaimed my presentation of a case of Candida-associated gastric ulcer at DDW 2012 in San Diego.
Conflict of Interests
I declare that I have no conflict of interests concerning this article.
References
- Cohen R, Roth FJ, Delgado E, Ahearn DG, Kalser MH. Fungal flora of the normal human small and large intestine. N Engl J Med. 1969; 280: 638-641.
- Eras P, Goldstein MJ, Sherlock P. Candida infection of the gastrointestinal tract. Medicine. 1972; 51: 367-379.
- Trier JS, Bjorkman DJ. Esophageal, gastric, and intestinal candidiasis. Am J Med. 1984; 77: 39-43.
- Minoli G, Terruzzi V, Butti G, Frigerio G, Rossini A. Gastric candidiasis: an endoscopic and histological study in 26 patients. Gastrointest Endosc. 1982; 28: 59-61.
- Katzenstein AA, Maksem J. Candida infection of gastric ulcers. Histology, incidence, and clinical significance. Am J Clin Pathol. 1979; 71: 137-141.
- Neeman A, Avidor I, Kadish U. Candidal infection of benign gastric ulcers in aged patients.” Am J Gastroenterol. 1981; 75: 211-213.
- Vilotte J, Toutoungi M, Coquillard A. Ulceres gastriques colonises par Candida. Caracteristiques cliniques et biologiques. Candida infection of gastric ulcers. 6 cases. Nouv Presse Med. 1981; 10: 1471-1474.
- Scott BB, Jenkins D. Gastro-oesophageal candidiasis. Gut. 1982; 23: 137- 139.
- Minoli G, Terruzzi V, Ferrara A, Casiraghi A, Rocca F, Rainer H, et al. A prospective study of relationships between benign gastric ulcer, Candida, and medical treatment. Am J Gastroenterol. 1984; 79: 95-97.
- Di Febo G, Miglioli M, Calo G, Biasco G, Luzza F, Gizzi G, et al. Candida albicans infection of gastric ulcer. Frequency and correlation with medical treatment. Results of a multicenter study. Dig Dis Sci. 1985; 30: 178-181.
- Morishita T, Kamiya T, Munakata Y, Tsuchiya M. Radiologic and endoscopic studies of gastric ulcers associated with Candida infection. Acta Gastroenterol Latinoam. 1993; 23: 223-229.
- Gottlieb-Jensen K, Andersen J. Occurrence of Candida in gastric ulcers. Significance for the healing process. Gastroenterology. 1983; 85: 535-537.
- Sasaki K. Candida-associated gastric ulcer relapsing in a different position with a different appearance. World J Gastroenterol. 2012; 18: 4450-4453.
- Brabander JOW, Blank F, Butas CA. Intestinal moniliasis in adults. Can Med Assoc J. 1957; 77: 478-483.
- Feng YW, Wu M, Li Y, Zeng JJ, Li ML, He Y, et al. Significance of identification of fungi in gastric juice of patients with artificial airway in intensive care unit. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2012; 24: 96-99.
- Goenka MK, Kochhar R, Chakrabarti A, Kumar A, Gupta O, Talwar P, et al. Candida overgrowth after treatment of duodenal ulcer. A comparison of cimetidine, famotidine, and omeprazole. J Clin Gastroenterol. 1996; 23: 7-10.
- Zwolinska-Wcislo M, Brzozowski T, Mach T, Trojanowska D, Konturek PC, Raido R, et al. Are probiotics effective in treatment of fungal colonization of the gastrointestinal tract? Experimental and clinical study. J Physiol Pharmacol. 2006; 57: 35-49.
- Brzozowski T, Zwolinska-Wcislo M, Konturek PC, Kwiecien S, Drozdowicz D, Knturek SJ, et al. Influence of gastric colonization with Candida albicans on ulcer healing in rats: effect of ranitidine, aspirin and probiotic therapy. Scand J Gastroenterol. 2005; 40: 286-296.
- Kumamoto CA. Inflammation and gastrointestinal Candida colonization. Curr Opin Microbiol. 2011; 14: 386-391.
- Sasaki K. Candida-associated gastric ulcer until yesterday, today, and from tomorrow --- In quest of the etiology ---. SciTz Gynecol Reprod Med. 2017; 1: 1002.
- Hirasaki S, Koide N, Ogawa H, Tsuji T. Benign gastric ulcer associated with Candida infection in a healthy adult. J Gastroenterol. 1999; 34: 688-693.
- Nishimura S, Nagata N, Kobayakawa M, Sako A, Nakashima R, Uemura N. A case of candidal infection of gastric ulcers with characteristic endoscopic findings. Nihon Shokakibyo Gakkai Zasshi. 2011; 108: 1393-1398.
- Zelante T, de Luca A, Bonifazzi P, Montagnoli C, Bozza S, Moretti S, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunology. 2007; 37: 2695-2706.
- Jacobsen ID, Wilson D, Wachtler B, Brunke S, Naglik JR, Hube B. Candida albicans dimorphism as a therapeutic target. Expert Rev Anti Infect Ther. 2012; 10: 85-93.
- Wachtler B, Citiulo F, Jablowski N, Forster S, Dalle F, Schaller M, et al. Candida albicans-epithelial interactions: dissecting the roles of active penetration, induced endocytosis and host factors on the infection process. PLoS One. 2012; 7: e36952.
- Silva S, Hooper SJ, Henriques M, Oliveira R, Azeredo J, Williams DW. The role of secreted aspartyl proteinases in Candida tropicalis invasion and damage of oral mucosa. Clin Microbiol Infect. 2011; 17: 264-272.
- Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj D, et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe. 2010; 8: 225-235.
- Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, et al. Human epithelial cells establish direct antifungal defense through TLR4- mediated signaling. J Clin Invest. 2007; 117: 3664-3672.
- Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature. 2016; 532: 64-68.
- Richardson JP, Mogavero S, Moyes DL, Blagojevic M, Kruger T, Verma AH, et al. Processing of Candida albicans Ece1p is critical for candidalysin maturation and fungal virulence.” MBio. 2018; 9: e02178-17.
- Ho J, Yang X, Nikou S-A, Kichik N, Donkin A, Ponde NO, et al. Candidalysin activates innate epithelial immune responses via epidermal growth factor receptor. Nat Commun. 2019; 10: 2297.
- Moyes DL, Murciano C, Runglall M, Islam A, Thavaraj S, Naglik JN. Candida albicans yeast and hyphae are discriminated by MAPK signaling in vaginal epithelial cells. PLoS One. 2011; 6: e26580.
- Moyes DL, Murciano C, Runglall M, Kohli A, Islam A, Naglik JR. Activation of MAPK/c-Fos induced responses in oral epithelial cells is specific to Candida albicans and Candida dubliniensis hyphae. Med Microbiol Immunol. 2012; 201: 93-101.
- Roselletti E, Perito S, Gabrielli E, Mencacci A, Pericolini E, Sabbatini S, et al. NLRP3 inflammasome is a key player in human vulvovaginal disease caused by Candida albicans.” Sci Rep. 2017; 7: 17877.
- Richardson JP, Willems HME, Moyes DL, Shoale S, Barker KS, Tan SL, et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa”. Infect Immun. 2018; 86: e00645- 17.
- Lee SA, Jones J, Khalique Z, Kot J, Alba M, Bernardo S, et al. A functional analysis of the Candida albicans homolog of Saccharomyces cerevisiae VPS4. FEMS Yeast Res. 2007; 7: 973-985.
- Lee SA, Jones J, Hardison S, Kot J, Khalique Z, Bernardo SM, et al. Candida albicans VPS4 is required for secretion of aspartyl proteases and in vivo virulence. Mycopathologia. 2009; 167: 55-63.
- Rane HS, Hardison S, Botelho C, Bernardo SM, Wormley F, Lee SA. Candida albicans VPS4 contributes differentially to epithelial and mucosal pathogenesis. Virulence. 2014; 5: 810-818.
- Vautier S, Drummond RA, Chen K, Murray GI, Kadosh D, Brown AJP, et al. Candida albicans colonization and dissemination from the murine gastrointestinal tract: the influence of morphology and Th17 immunity. Cell Microbiol. 2015; 17: 445-450.
- Hernandez-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe. 2012; 11: 425-435.
- Vautier S, Drummond RA, Redelinghuys P, Murray GI, MacCallum DM, Brown G. Dectin-1 is not required for controlling Candida albicans colonization of the gastrointestinal tract. Infect Immun. 2012; 80: 4216-4222.
- Dalle F, Wachtler B, L’Ollivier C, Holland G, Norbert B, Wilson D, et al. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 2010; 12: 248-271.
- Kavanaugh NL, Zhang AQ, Nobile CJ, Johnson AD, Ribeck K. Mucins suppress virulence traits of Candida albicans. MBio. 2014; 5: e01911.
- Colina AR, Aumont F, Deslauriers N, Belhumeur P, de Repentigny L. Evidence for degradation of gastrointestinal mucin by Candida albicans secretory aspartyl proteinase. Infect Immun. 1996; 64: 4514-4519.
- Goyer M, Loiselet A, Bon F, L’Ollivier C, Laue M, Holland G, et al. Intestinal cell tight junctions limit invasion of Candida albicans through active penetration and endocytosis in the early stages of the interaction of the fungus with the intestinal barrier. PLoS One. 2016; 11: e0149159.
- Liu Y, Filler SG. Candida albicans Als3, a multifunctional adhesin and invasion. Eukaryotic Cell. 2011; 10: 168-173.
- Bohringer M, Pohlers S, Schulze S, Albrecht-Eckardt D, Piegsa J, Weber M, et al. Candida albicans infection leads to barrier breakdown and a MAPK/NF- κB mediated stress response in the intestinal epithelial cell line C2BBe1. Cell Microbiol. 2016; 18: 889-904.
- Wachtler B, Wilson D, Haedicke K, Dalle F, Hube B. From attachment to damage: defined genes of Candida albicans mediate adhesion, invasion and damage during interaction with oral epithelial cells. PLoS One. 2011; 6: e17046.
- Allert S, Forster TM, Svensson C-M, Richardson JP, Pawlik T, Hebecker B, et al. Candida-albicans-induced epithelial damage mediates translocation through intestinal barriers. MBio. 2018; 9: e00915-18.
- Los FCO, Randis TM, Aroian RV, Ratner AJ. Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev. 2013; 77: 173-207.
- Willems HME, Lowes DJ, Barker KS, Palmer GE, Peters BM. Comparative analysis of the capacity of the Candida species to elicit immunopathology. Infect Immun. 2018; 86: e00527-18.
- Hernandez-Santos N, Huppler AR, Peterson AC, Khader SA, McKenna KC, Gaffen SL. Th17 cells confer long term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013; 6: 900-910.
- Richardson JP, Moyes DL. Adaptive immune responses to Candida albicans infection. Virulence. 2015; 6: 327-337.
- Tso GHW, Reales-Calderon JA, Tan ASM, Sem X, Le GTT, Tan TG, et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science. 2018; 362: 589-595.
- Bistoni F, Vecchiarelli A, Cenci E, Puccetti P, Marconi P, Cassone A. Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infect Immun. 1986; 51: 668-674.
- Bistoni F, Verducci G, Perito S, Vecchiarelli A, Pucceti P, Marconi P, et al. Immunomodulation by a low-virulence, agerminative variant of Candida albicans. Further evidence for macrophage activation as one of the effector mechanisms of nonspecific anti-infectious protection. J Med Vet Mycol. 1988; 26: 285-299.
- Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocyte. Cell Host Microbe. 2012; 12: 223- 232.
- Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science. 2014; 345: 1251086.
- Ifrim DC, Quintin J, Joosten LAB, Jacobs C, JansesnT, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. 2014; 21: 534-545.
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR/HIF1a-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014; 345: 1250684.
- Alburquenque C, Amaro J, Fuentes M, Falconer MA, Moreno C, Covarrubias C, et al. Protective effect of inactivated blastoconidia in keratinocytes and human reconstituted epithelium against C. albicans infection. Med Mycol. 2019; 57: 457-467.
- Yano J, Lilly E, Barousse M, Fidel PL. Epithelial cell-derived S100 calciumbinding protein as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun. 2010; 78: 5126-5137.
- Gao Y, Liang G, Wang Q, She X, Shi D, Shen Y, et al. Different host immunological response to C. albicans by human oral and vaginal epithelial cells. Mycopathologia. 2019; 184: 1-12.
- Korn T, Betelli E, Oukka M, Kuchroo V. IL-17 and Th17 Cells. Annu Rev Immunol. 2009; 27: 485-517.
- Lilic D. Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol. 2012; 15: 420-426.
- von Bernuth H, Picard C, Puel A, Casanova J-L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunnol. 2012; 42: 3126-3135.
- Vogelaar IP, Ligtenberg MJL, van der Post RS, de Voer RM, Kets CM, Jansen TJG, et al. Recurrent candidiasis and early onset gastric cancer in a patient with a genetically defined partial MYD88 defect. Fam Cancer. 2016; 15: 289-296.
- Conti HR, Gaffen SL. IL-17-mediated immunity to the opportunistic fungal pathogen Candida albicans. J Immunol. 2015; 195: 780-788.
- Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol. 2017; 2: eaam8834.
- Bishu S, Hernandez-Santos N, Simpson-Abelson MR, Huppler AR, Conti HR, Ghilardi N, et al. The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun. 2014; 82: 1173-1180.
- Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor FEY, et al. IL- 36 and IL-1/IL-17 drive immunity to oral candidiasis via parallel mechanisms. J Immunol. 2018; 201: 627-634.
- Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. 2013; 190: 521-525.
- Conti HR, Peterson AC, Brane L, Huppler AR, Hernández-Santos N, Whibley N, et al. Oral-resident natural Th-17 cells and γδ T cells control opportunistic Candida albicans infections. J Exp Med. 2014; 211: 2075-2084.