
Abstract
Background: Endurance exercise races have grown in popularity over the last several years. Even if regular exercise is beneficial for cardiovascular health, many studies also demonstrated that running a marathon or a longdistance triathlon induces transient Left Ventricle (LV) dysfunctions associated in some athletes with myocardial damages. The underlying mechanisms of these ventricular dysfunctions are not fully elucidated.
Methods: Adult male Wistar rats were randomly assigned to the following 4 groups (8 rats per group): Control (Ctrl), Exercised Group (4 hours run on treadmill) (Ex) and 2 groups in which NADPH oxidase enzyme was inhibited with apocynin treatment (Ctrl-APO and Ex-APO, respectively). Cardiac function was evaluated ex vivo during basal conditions using the isolated perfused Langendorff heart model. Connexin 43 (Cx43) dephosphorylation was assessed in LV myocardium by western blotting.
Results: The prolonged exercise of 4 hours on treadmill induced a decrease in LV intrinsic myocardial contractility and relaxation and a dephosphorylation of Cx43 in the LV myocardium. Interestingly, the apocynin treatment, specific inhibitor of the NADPH enzyme, reversed LV dysfunctions and prevented Cx43 dephosphorylation.
Conclusion: The present findings show a potential implication of the NADPH oxidase enzyme in myocardial connexin 43 dephosphorylation which can be involved in LV dysfunctions following a prolonged exercise in rat. This new underlying mechanism insight of exercise-induced cardiac fatigue may help to determine the long-term effects of prolonged exercises on cardiac risk in athletes.
Keywords: Cardiac fatigue; Connexin 43; Oxidative stress; Prolonged exercise
Abbreviations
cTnI: Cardiac Troponin I; Ctrl: Control Group; Ctrl-APO: Control Group Treated with apocynin; Cx43: Connexin 43; dP/ dtmax: Left Ventricle Intrinsic Myocardial Contractility; dP/dtmin: Relaxation; Ex: Exercised Group; Ex-APO: Exercised Group Treated with apocynin; LV: Left Ventricle; NADPH: Nicotinamide Adenine Dinucleotide Phosphate Oxidase Enzyme; PSE: Prolonged Strenuous Exercise
Introduction
The popularity of endurance exercise races has increased worldwide over the last thirty years. However, a growing number of studies also demonstrated that running a long-distance race induces transient ventricular dysfunctions associated in some subjects with an increase in Cardiac Troponin I (cTnI), a biomarker of myocardial damages [1,2]. Oxidative stress has been proposed to explain this endurance exercise-induced cardiac fatigue [3,4]. Among the potential sources of oxidative stress like xanthine oxidase, uncoupled nitric oxide synthases and the mitochondrial electron transport chain, the Nicotinamide Adenine Dinucleotide Phosphate Oxidase Enzyme (NADPH) is considered as major sources of ROS in the myocardium [5,6]. Our research team has previously reported a significant link between NADPH oxidase-dependent oxidative stress and myocardial dysfunction after a Prolonged Strenuous Exercise (PSE) on treadmill in rat [3]. Despite some advances in the explanation of exerciseinduced cardiac fatigue, it was recently mentioned that mechanisms producing the decrease in cardiac function after exercise still not clear because of the multiplicity of factors which may contribute to this phenomenon [7]. Gap junctions are essential for the heart function notably by their important role in impulse conduction. They are low resistance connections between adjacent myocardial cells and are predominantly composed by the connexin 43 (Cx43) [8]. In addition, the phosphorylation of Cx43 is essential for gap junction plaque forming and thus for intercellular communication [9-11]. Previous studies in animals have demonstrated that pathological conditions like heart failure and cardiac ischemia induce Cx43 protein dephosphorylation which regulate connexin function [12- 14]. Furthermore, Cx43 dephosphorylation can be induced by oxidative stress generated by the NADPH oxidase enzyme which can exhert arrhythmogenic effect [12]. Therefore, among the potential mechanisms, gap junctions remodeling might be involved in PSEinduced LV dysfunctions.
However, to date, the impact of PSE on NADPH oxidasedependent oxidative stress on Cx43 phosphorylation status associated with LV myocardial function assessment have not been previously described. Thus, the hypothesis of the present study is that NADPH oxidase-dependent redox status modulation induces Cx43 dephosphorylation which represents one of the underlying mechanisms involved in LV myocardial dysfunctions after a PSE.
Methods
All procedures were performed in agreement with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publications No. 85-23, revised 1996) and approved by the French ministry of agriculture. Adult Wistar male rats were randomly assigned to the following groups (8 rats per group): one Control Group (Ctrl) and one Exercised Group (Ex). Two other groups of 8 rats were treated with apocynin, (Sigma Aldrich, France) a bio-absorbable specific inhibitor of the NADPH oxidase enzyme, in drinking water (1.5 mM) as previously described [15] and were assigned to a Ctrl-APO and Ex-APO groups. All exercise sessions were performed on a motorized rodent treadmill. All exercised animals were first familiarized with the treadmill over 5 days then performed an incremental test to determine their maximal running speed and finally performed a Prolonged Strenuous Bout of Exercise (PSE) at 60-65% of their maximal running speed for 4 hours.
All the Ctrl, Ctrl-APO (n = 8 / group) and Ex, Ex-APO (n = 8 / group) animals were killed within 30 min following exercise for isolated heart experiments as previously described [3]. Another set of animals (Ctrl, Ctrl-APO and Ex, Ex-APO; n = 4 / group) were killed within 30 min following exercise for western blots analyses of the nonphosphorylated form (i.e. P0 form) of the Cx43 protein (monoclonal Cx43 antibody (13-8300), Thermo Fisher Scientific, France) [16].
Statistical Analysis
Data were analyzed using one-way or two-way ANOVA among groups. When significant interactions were found, a Bonferroni t-test was applied with adjusted P>0.05 (Stat view 2.20; Adept Scientific, Letch worth, UK). Data are presented as means ± S.E.M.
Results
The cardiac function of Ex animals was altered by the PSE compared to Ctrl animals (Figure 1). The LV intrinsic myocardial contractility (dP/dtmax) and relaxation (dP/dtmin) were respectively significantly decreased by 37% (P>0.05) and 34% (P>0.05) in Ex rats compared to Ctrl rats. The specific inhibition of NADPH oxidase by the apocynin treatment improved myocardial function in Ex-APO rats to the level of Ctrl and Ctrl-APO rats (Figure 1). Similarly, LV dP/dtmax (Figure 1, upper graph), dP/dtmin (Figure 1, lower graph) were normalized in Ex-APO rats to the level found in Ctrl and Ctrl- APO rats. The PSE induced a 41% increase of the non-phosphorylated status of Cx43 in Ex animals compared to Ctrl animals (P>0.05, Figure 2). Interestingly, the apocynin treatment normalized Cx43 phosphorylation status in Ex-APO rats to the level found in Ctrl and Ctrl-APO rats (Figure 2).
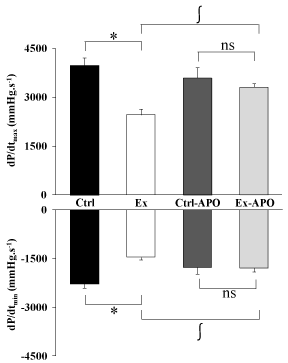
Figure 1: Effect of a 4-hour treadmill run and apocynin treatment on left
ventricular function in control and exercised rats.
Ex-vivo cardiac function in rat. Graphs depict the left ventricle contractility
(dP/dtmax - upper graph) and relaxation (dP/dtmin - lower graph) in Ctrl: control
rats; Ex: exercised rats; Ctrl-APO: control rats with apocynin treatment; Ex-
APO: exercised rats with apocynin treatment measured using Langendorff
isolated and perfused heart model after PSE. Values are mean ± S.E.M. (n =
8/group). * P<0.05 vs. control; ∫ P<0.05 vs. apocynin.
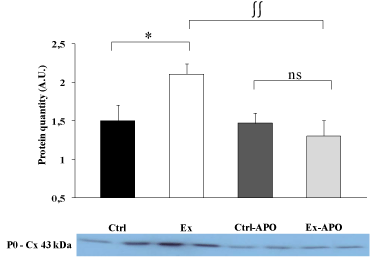
Figure 2: Effect of a 4-hour treadmill run and apocynin treatment on
myocardial connexin 43 dephosphorylation in the myocardium of
control and exercised rats.
Western blots data for the myocardial non-phosphorylated form of the
connexin 43 protein (i.e. P0 form) in the left ventricle myocardial tissue of
Ctrl: control rats; Ex: exercised rats; Ctrl-APO: control rats with apocynin
treatment; and Ex-APO: exercised rats with apocynin treatment. Cx 43,
connexin 43. Values are mean ± S.E.M. (n = 4/group). * P<0.05 vs. control;
∫∫ P<0.01 vs. apocynin.
Discussion
Prolonged and ultra-endurance exercises have been related to a decrease in cardiac function in active healthy subjects and athletes [17,18]. This phenomenon called exercise-induced cardiac fatigue was also observed in animals [3,19]. In a very recent review, authors addressed all of the known adverse cardiovascular effects of endurance exercise [7]. They concluded that although regular physical activity and exercise training is highly beneficial for the population health, prolonged exercise can adversely affect cardiac function in some subjects. Most importantly, these authors mentioned that the mechanisms producing the decrease in cardiac function after exercise still not clear because of the multiplicity of factors which may contribute to this phenomenon.
Some studies have been done to better understand the potential underlying mechanisms of this cardiac dysfunction induced by PSE. Authors have demonstrated myocardial damage in rat after 3 hours of swimming with 5% of the body weight workload [4,19]. Our team has also demonstrated that PSE induces impairment in intrinsic myocardial function and that NADPH oxidase-induced modification of redox status was a potential new trigger of PSEinduced cardiac dysfunction [3]. The findings of the present study showed that Cx43 dephosphorylation appeared in myocardial tissue of rats after PSE and that apocynin treatment normalized the level of myocardial Cx43 phosphorylation after a PSE in the Ex group. These results are in line with a previous study where oxidative stress has been suggested to be involved in Cx43 dephosphorylation during myocardial ischemia/reperfusion in rat [20]. This phenomenon may be involved in cardiac rhythm perturbations as previously reported [12-14] and ultimately in LV dysfunctions. The present findings are also in line with a previous study where the total Cx43 mRNA, protein expression and the phosphorylated Cx43 protein were decreased in rabbits with heart failure and where apocynin treatment completely reversed this reduction and induced antiarrhythmogenic effects [12]. Moreover, cardiac adrenoreceptors are directly involved in the control of intercellular electrical communication and probably critical for the maintenance of regular cell-to-cell conduction throughout the myocardium [21]. Thus, PSE might induce a sort of chronic adrenergic stimulation of non-phosphorylated Cx43 which may probably worsen the formation of an arrhythmogenic substrate in the heart of exercised rats.
Conclusion
In conclusion, the results of the present study demonstrate one implication of the NADPH oxidase enzyme in LV dysfunctions following PSE in rat and suggest the implication of oxidative stress from this enzyme in Cx43 dephosphorylation. This new underlying mechanism might be one of the potential mechanisms involved in exercise-induced cardiac fatigue and may help to determine the longterm effects of prolonged exercises on cardiac risk in athletes.
Acknowledgement
I would like to thank all of the members of the LAPEC lab (EA4278) from the University of Avignon, France for their support in this work.
References
- Middleton N, Shave R, George K, Whyte G, Hart E, Atkinson G. Left ventricular function immediately following prolonged exercise: A meta-analysis. Med Sci Sports Exerc. 2006; 38: 681-687.
- Shave R, Baggish A, George K, Wood M, Scharhag J, White G, et al. Exercise-induced cardiac troponin elevation: evidence, mechanisms, and implications. J Am Coll Cardiol. 2010; 56: 169-176.
- Vitiello D, Boissiere J, Doucende G, Gayrard S, Polge A, Faure P, et al. beta-Adrenergic receptors desensitization is not involved in exercise-induced cardiac fatigue: NADPH oxidase-induced oxidative stress as a new trigger. J Appl Physiol (1985). 2011; 111: 1242-1248.
- Nie J, Close G, George KP, Tong TK, Shi Q. Temporal association of elevations in serum cardiac troponin T and myocardial oxidative stress after prolonged exercise in rats. Eur J Appl Physiol. 2010; 110: 1299-1303.
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004; 4: 181-189.
- Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006; 71: 208-215.
- Eijsvogels TM, Fernandez AB, Thompson PD. Are There Deleterious Cardiac Effects of Acute and Chronic Endurance Exercise? Physiol Rev. 2016; 96: 99-125.
- Scemes E, Spray DC, Meda P. Connexins, pannexins, innexins: novel roles of "hemi-channels". Pflugers Arch. 2009; 457: 1207-1226.
- Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991; 273: 67-72.
- Musil LS, Good enough DA. Biochemical analysis of connexin 43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991; 115: 1357-1374.
- Solan JL, Lampe PD. Connexin 43 phosphorylation: structural changes and biological effects. Biochem J. 2009; 419: 261-272.
- Liu Y, Huang H, Xia W, Tang Y, Li H, Huang C, et al. NADPH oxidase inhibition ameliorates cardiac dysfunction in rabbits with heart failure. Mol Cell Biochem. 2010; 343: 143-153.
- Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000; 87: 656-662.
- Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, et al. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. 2004; 95: 726-733.
- Yamamoto E, Lai ZF, Yamashita T, Tanaka T, Kataoka K, Takutomi Y, et al. Enhancement of cardiac oxidative stress by tachycardia and its critical role in cardiac hypertrophy and fibrosis. J Hypertens. 2006; 24: 2057-2069.
- Nagy JI, Li WE, Roy C, Doble BW, Gilchrist JS, Kardami E, et al. Selective monoclonal antibody recognition and cellular localization of an unphosphorylated form of connexin 43. Exp Cell Res. 1997; 236: 127-136.
- Vitiello D, Cassirame J, Menetrier A, Rupp T, Schuster I, Reboul C, et al. Depressed systolic function after a prolonged and strenuous exercise. Med Sci Sports Exerc. 2013; 45: 2072-2079.
- Vitiello D, Rupp T, Bussiere JL, Robach P, Polge A, Millet GY, et al. Myocardial damages and left and right ventricular strains after an extreme mountain ultra-long duration exercise. Int J Cardiol. 2013; 165: 391-392.
- Olah A, Nemeth BT, Matyas C, Horvath EM, Hidi L, Birtalan E, et al. Cardiac effects of acute exhaustive exercise in a rat model. Int J Cardiol. 2015; 182: 258-266.
- Rakotovao A, Tanguy S, Toufektsian MC, Berthonneche C, Ducros V, Tosaki A, et al. Selenium status as determinant of connexin-43 dephosphorylation in ex vivo ischemic/reperfused rat myocardium. J Trace Elem Med Biol. 2005; 19: 43-47.
- Salameh A, Dhein S. Adrenergic control of cardiac gap junction function and expression. Naunyn Schmiedebergs Arch Pharmacol. 2011; 383: 331-346.