
Abstract
Identifying all the molecular components within a living cell is the first step into understanding how it functions. To further understand how a cell functions requires identifying the interactions that occur between these components. This fact is especially relevant for proteins. No protein within a human cell functions on its own without interacting with another biomolecule - usually another protein. While Protein-Protein Interactions (PPI) have historically been determined by examining a single protein per study, novel technologies developed over the past couple of decades are enabling high-throughput methods that aim to describe entire protein networks within cells. In this review, some of the technologies that have led to these developments are described along with applications of these techniques. Ultimately the goal of these technologies is to map out the entire circuitry of PPI within human cells to be able to predict the global consequences of perturbations to the cell system. This predictive capability will have major impacts on the future of both disease diagnosis and treatment.
Keywords: Protein-protein interactions; Mass spectrometry; Affinity purification; Cross-linking; Proximity labeling
Introduction
Living cells are the ultimate team. To function properly, the players on the cell’s team (i.e., DNA, RNA, proteins, metabolites, etc.) must interact with each other at the correct location and proper time [1]. These interactions drive every cell function. For DNA to be properly replicated or transcribed, it must interact with proteins, RNA, and metabolites. For RNA to be translated into proteins, it must interact with proteins and other RNA molecules. Besides DNA replication, RNA transcription, and protein translation, biomolecular interactions maintain the cell’s structure (e.g., actin filaments), transport molecules throughout the cell, interpret and propagate signals originating from outside the cell (e.g., receptors, kinases, phosphatases, etc.), orchestrate cell division (e.g., cyclins, etc.), and produce the energy required for all of these processes to occur.
While interactions between diverse groups of biomolecules are critical to cell function, understanding Protein-Protein Interactions (PPI) are especially important to decipher. For example, if a novel protein is discovered, identifying who it interacts with provides a key piece of information for determining its function. Basic research has long recognized the importance of identifying PPI as illustrated by the large number of manuscripts on this topic that are published in top tier journals. Historically, hypothesis-driven methods have been used to identify suspected PPI [2-4]. In many hypothesis-driven methods, cells are lysed under non-denaturing conditions to preserve PPI as much as possible outside of their native environment (Figure 1A). A targeted protein is captured, along with other biomolecules that are bound to the target protein, using an affinity device (usually an antibody). A series of washing steps are performed to eliminate nonspecifically bound proteins, while retaining those that are legitimate members of the protein complex. After separating the members of the protein complex using Polyacrylamide Gel Electrophoresis (PAGE), they are transferred to a Polyvinylidene Fluoride (PVDF) membrane. This stage is where the hypothesis comes in. The PVDF membrane is probed with an antibody that targets a protein that the investigator believes is part of the complex. If the antibody reveals a band near the anticipated molecular weight of the hypothesized protein, it is concluded that this protein interacts with the target protein. The net result is binary discovery: a second protein that interacts with the target protein is discovered [5].
While this hypothesis-driven method has proven fruitful, there are several deficiencies in this approach. Since it requires using a specific antibody probe to prove a hypothesis, incorrect hypotheses can be costly in terms of both time and money. Unexpected, novel PPI are difficult to find using this strategy. The technique is heavily reliant on antibody specificity for identifying novel PPI. Regardless of these deficiencies, hypothesis-driven methods continue to play a major role in basic research [6].
The advent of modern Mass Spectrometry (MS) technologies has made a huge impact on the identification of PPI. The characterization of PPI is arguably the biggest impact MS has had on biological sciences; even greater than its role in systems biology or biomarker discovery. There are a number of reasons for this impact. Firstly, MS has shifted PPI studies from hypothesis to discovery-driven [7]. While the sample preparation steps (i.e., isolation of the protein complex) are similar, the discovery-driven method differs in how and how many proteins can be identified in a single study. In a discovery-driven approach, the isolated complex is fractionated (generally using either SDS-PAGE or liquid chromatography) and all of the proteins present are analyzed using MS (Figure 1B). The advantages of this method are that no hypothesis is needed to identify interacting proteins and multiple members of the protein complex can be identified without the need for antibodies. Another advantage of this discovery-driven approach is that proteins that could never have been predicted to be part of the targeted complex can be identified. Finally, since protein identification is not reliant on antibodies there are no issues related to the uncertainty associated with antibody cross-reactivity.
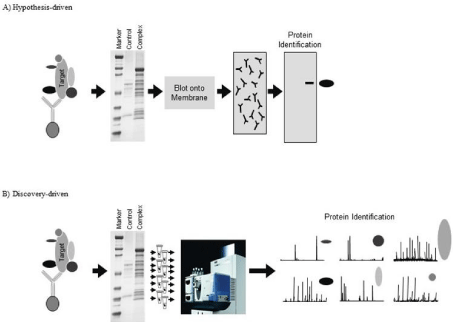
Figure 1: Discovery and hypothesis-driven methods of identifying Protein-Protein Interactions (PPI). A) In a hypothesis-driven approach, a target protein and its
binding partners is extracted from cells using techniques such as immunoprecipitation. The extracted proteins are fractionated using gel electrophoresis and then
blotted onto a membrane. This membrane is probed with an antibody specific for a protein that is hypothesized to interact with the target protein. B) The discoverydriven
approach uses identical protein complex extraction methods; however, the gel-fractionated and stained proteins are extracted individually from the gel.
These proteins (target and binding proteins) are then identified using MS.
Whole Proteome Screening Using Tandem Affinity Purification and Mass Spectrometry
Discovery-driven methods for characterizing PPI have begun to supplant hypothesis-driven methods for many of the reasons described above. As with evolving technologies, some scientists saw the ability to identify multiple PPI in a single study as an opportunity to generate an entire protein network for the cell. The hope was that by generating an entire network of PPI a predictive capability of what happens when the cell is perturbed (either naturally or artificially) could be developed.
The initial attempt at generating a cell-wide PPI map involved two studies using Saccharomyces cerevisiae (S. cerevisiae) as a model organism [8,9]. S. cerevisiae has a molecular architecture similar to mammalian cells and the function of many human proteins were identified through the discovery of their homolog in yeast. In addition, S. cerevisiae can be easily genetically manipulated and transformed to express chimeric proteins necessary to identify hundreds of protein complexes [10].
Both studies used a strategy of labeling specific protein targets with the polypeptide tag FLAG [8] or a dual tag comprised of both Protein A and Calmodulin Binding Peptide (CBP) [9]. These labeling strategies enabled the use of a single type of antibody to extract the various protein complexes. Both strategies used SDSPAGE fractionation followed by MS identification of the separated proteins. The study that utilized Protein A/CBP-tagging reported 491 complexes comprised of 1,483 proteins [9], while the study that used FLAG-tagging reported 547 complexes containing 2,702 proteins [8]. While both studies provided impressive results, concern was raised when the two studies were compared. Only about 33% of the proteins were identified in both studies [11]. In general, the overlap in proteins identified in most of the complexes in either study was less than 50%.
While the overlap between these two studies was low, it could be explained by the number of non-specific interactions that are often observed in such studies. For instance, both studies used a single protein complex isolation method for all target proteins. Owing to their range of affinities, the isolation conditions must be carefully optimized for each complex to maximize the number of specifically bound proteins while minimizing the number of non-specifically bound ones. While the results ultimately did not produce an accurate protein network of a yeast cell, they accomplished something more important: they planted the seeds for further investigations into cellwide PPI.
Techniques for Analyzing Multi-Protein Complexes
The ability to even consider identifying protein complexes originated with the development of MS technologies for identifying thousands of proteins in a high-throughput manner [12,13]. Once this technology had sufficiently matured, many investigators turned their attention to sample preparation methods that would allow not only PPI to be identified, but also networks of PPI. While there have been a number of different techniques developed for identifying PPI (i.e., yeast-two hybrid screens, protein arrays, etc.), I am going to focus on techniques that specifically incorporate downstream MS analysis.
Affinity Pulldown Mass Spectrometry
Affinity pulldown (AP) methods represent a straightforward approach for identifying PPI as they rely on well-established techniques for both the sample preparation and MS analysis [14-17]. The major steps required for AP-MS analysis include; i) expressing an epitope-tagged target protein within the cells; ii) immunoprecipitating the tagged protein using an antibody directed against the epitope tag; and iii) analyzing the extracted complex using MS (Figure 2A). With the commercial availability of a large number of open-reading frames, a vast selection of protein targets can be tagged with various epitopes (i.e., FLAG, CBP, TAP, etc.) enabling PPI data for a very large number of proteins to be determined. The method is relatively high-throughput as only one type of antibody (directed against the specific tag) is required for isolating the protein complexes.
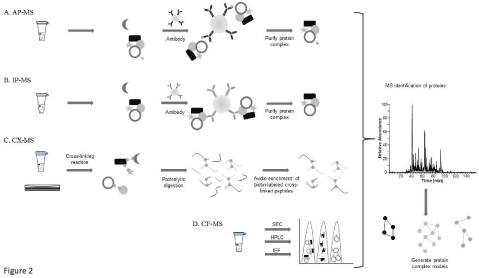
Figure 2: Schematic of methods used to characterize Protein-Protein Interactions (PPI) using A) Affinity-Purification Mass Spectrometry (AP-MS), B)
Immunoprecipitation Mass Spectrometry (IP-MS), C) Cross-Linking Mass Spectrometry (CX-MS), and D) Co-Fractionation Mass Spectrometry (CF-MS).
Immunoprecipitation Mass Spectrometry
Since the Immunoprecipitation (IP) method, as described earlier in this article, is very similar to the AP-MS method it will only be discussed briefly. In the IP-MS method, an antibody is used to isolate a target protein under non-denaturing conditions so that other proteins bound to this target protein will also be extracted from the cell lysate (Figure 2B) [18-20]. The isolated complex is then characterized using MS. Like AP-MS, the IP-MS method is relatively high-throughput, but requires a specific antibody for each target protein.
Cross-Linking Mass Spectrometry
Cross-linking of proteins followed by MS analysis (CX-MS) utilizes cross-linking reagents that form covalent bonds between proteins in close proximity [24-26]. The crosslinking step can be performed either in vivo or in vitro (Figure 2C). The in vivo method links the proteins together within their native cell environment, thereby minimizing the number artifactual interactions and loss of positive interactions resulting from ex vivo sample preparation steps. In the in vitro method, the cross-linking reagent is added to the protein lysate after it has been extracted from the cell using nondenaturing conditions. The lysate is then digested into peptides and cross-linked peptides are extracted from the mixture and identified using LC-MS. Unlike the IP-, AP-, and Co-Fractionation (CF)-MS (described below) methods, CX-MS provides data related to the precise structural site of interaction between two proteins. CX-MS does not require protein-specific antibodies and there are a wide range of chemical crosslinking reagents available enabling the study of short- or long-range PPI.
Co-Fractionation Mass Spectrometry
An emerging method for characterizing PPI is biochemical cofractionation followed by MS analysis (CF-MS) [21-23]. Just like APMS and IP-MS, this method requires samples be maintained under non-denaturing conditions during the sample preparation phase. In CF-MS, the cell lysate is fractionated using different chromatographic techniques including size exclusion, ion exchange, or hydrophobic interaction (Figure 2D). The separated fractions are then analyzed using MS. The PPI are assumed based on the co-elution of proteins within the final fractions along with bioinformatic analysis of the data that determines the relevance of the functional interaction (i.e., does the interaction have any precedence in literature or can a functional basis for the interaction be postulated). The major advantage of the CF-MS method is its universal applicability to all types of biological samples and it does not require protein tagging or antibody purification.
Challenges in AP-, IP-, CX- and CF-MS Analyses
Like most analyses, characterizing PPI obeys the refrain of GIGO (Garbage-In, Garbage-Out). The most crucial step for the successful identification of PPI is the isolation of the complex. Identification of the components using MS is non-biased; therefore, it identifies any protein to which it is introduced regardless of its specificity within the complex. Each step needs to be carefully conducted with the aim of isolating the protein complex as it would exist in vivo. The initial step of lysing the cells or tissue must be done under sufficiently harsh conditions to allow access to the protein complexes, but not so harsh that complexes do not remain intact. If the complex extraction procedure is not optimized to minimize non-specifically bound proteins, these will be identified along with specifically bound proteins. In discovery-driven studies, this type of promiscuous result makes it very difficult to generate any reliable conclusions concerning functional protein interactions. Conversely, if the complex is isolated using conditions that are too harsh, weakly bound proteins will be lost. In this case, many proteins that functionally interact with the target protein will be lost prior to MS analysis.
The techniques described previously have proven to be enormously successful in determining PPI, but they are not without their own specific deficiencies [27]. All of the methods can result in a high false discovery rate, requiring each step in the process to be carefully optimized. Even under optimized conditions, non-specifically bound proteins will be observed regardless of which technique is chosen. The AP-, IP-, and CF-MS methods are all biased against weak or transient interactions since in vivo conditions cannot be maintained throughout the sample preparation steps. Since the AP-MS method requires overexpression of the target protein, artifactual PPI are often observed based on the simple fact that the protein’s wild-type stoichiometry has been altered. The CX-MS method has a unique disadvantage not observed with the other three. To identify proteins with using the AP-, IP-, and CF-MS methods, the samples are digested into peptides that are identified using well-established MS methods. Since the CX-MS method incorporates a covalently bound tag that couples two peptides, identification of the peptides is more complicated. This complicity requires special algorithms to convert the raw MS data into the correct peptide sequences. A summary of the advantages and disadvantages of each technique in identifying PPI is provided in Table 1.
![]()
Method
Advantages
Challenges
AP-MS
- Relatively high-throughput.
- Large number of commercially available ORFs for tagging.
- Single optimization technique required for antibody extraction.
- MS identification straightforward.
- High false discovery rate.
- Over-expression of target protein leads to artifacts.
- Weak or transient interactions can be missed.
IP-MS
- Relatively high-throughput.
- MS identification straightforward.
- High false discovery rate.
- Conditions need to be optimized for each antibody extraction.
- Weak or transient interactions can be missed.
CF-MS
- Identifies native stable complexes.
- Protein tagging or expression not required.
- No antibodies required.
- Biased against weak or transient interactions.
- Requires a lot of protein sample and significant MS capabilities.
CX-MS
- Provides information related to sites of interaction.
- Various crosslinkers available.
- Protein tagging or expression not required.
- No antibodies required.
- Requires careful optimization of reaction conditions to minimize false positives.
- Identification of cross-linked peptides is challenging.
BLPL
- Identifies weak interactions.
- Identifies transient interactions.
- Requires protein tagging with enzyme.
- Tagging can alter target protein function.
- Low throughput.
APEX
- Captures transient interactions.
- Useful for capturing protein interactors in close proximity.
- Requires protein tagging with enzyme.
- Toxicity of reagent limits in vivo use.
- Low throughput.
Table 1: Advantages and challenges of identifying protein-protein interactions using Affinity Purification-Mass Spectrometry (AP-MS); Immunoprecipitation MS (IPMS); Co-Fractionation MS (CF-MS); Cross-Linking MS (CX-MS); Biotin-Ligase Proximity Labeling (BLPL) and Ascorbate Peroxidase (APEX) PL methods.
Proximity Labeling Methods
The speed by which some processes occur within the cell is astonishing. This pace requires on/off rates between protein interactions to be rapid. Detecting these transient interactions is not reliable using the PPI methods described above, but require techniques that can capture and preserve transient PPI. Such transient interactions are not optimally detected using AP-, IP-, and CF-MS methods. Another weakness of AP-, IP, and CF-MS methods is their bias against weak interactions. This bias is a result of the loss of weak interactions during the sample preparation and fractionation steps required prior to complex identification. Preserving weak and transient interactions requires techniques that can rapidly label physically adjacent proteins without creating a background of collaterally labeled proteins. The development of Proximity Labeling (PL) techniques have provided further opportunities for studying the dynamics of PPI in living cells [28]. Proximity labeling uses enzymes fused to target proteins that covalently modify proteins that are physically adjacent to the target protein. Most PL reagents will label proteins within a roughly 10 nm radius of the target protein to which the enzyme is fused. Proximity labeling techniques requires the engineering of chimeric proteins containing the target protein coupled with an enzyme such as biotin ligase, biotin peroxidase, or Ascorbate Peroxidase (APEX) [29-31].
Horseradish Peroxidase, Ascorbate Peroxidase, and Biotin Ligase Proximity Labeling
Proximity labeling using Horseradish Peroxidase (HRP) and APEX reagents couple HRP or APEX to a target protein. In the presence of an exogenous biotin-phenol reagent and H2O2, the chimeric protein will catalyze the addition of a phenoxyl-biotin radical to proteins within its immediate vicinity. Biotin ligase PL methods, which use a chimeric target protein coupled to biotin ligase, utilize intracellular ATP to couple biotin to proximal proteins. Once the biotin is coupled to the proteins, the cells are lysed and the biotin-labeled proteins are enriched using streptavidin. Proteins that are part of the target protein complex are identified using MS. Since the proteins are covalently coupled to biotin, denaturing cell lysis conditions can be used since it is not necessary to preserve the protein complex at this point. The HRP method is very well suited for cell surface and secretory proteins owing to the oxidative environment in these regions [32]. While the APEX method is rapid and produces a limited labeling radius, making it optimal for studying dynamic interactions, it does require treating cells with H2O2 that can perturb wild-type PPI. In addition, since the cell membrane is impermeable to the biotin-phenol reagent used in APEX labeling, this PL technique has limited capability for detecting cytoplasmic PPI.
Detecting weak and transient cytoplasmic PPI is more amenable using biotin ligases owing to the high cellular membrane permeability of biotin and the use of intracellular ATP for labeling [33]. Earlier versions of chimeric protein target/biotin ligases required lengthy labeling times (i.e., 24h) resulting in a loss of dynamic and transient interactions as well as increased background labeling of non-specific interactions [34]. Newer versions of the engineered proteins, however, have faster kinetics and lower affinity for biotin enabling a greater specificity for labeling transient PPI [35].
What are the deficiencies of these PL techniques? The most obvious deficiency is its requirement for engineered tagged proteins, similar to what is required for AP-MS. This requirement leads to its second major deficiency. While CF- and CX-MS can be used to conduct global PPI studies from a single sample, PL methods can only analyze a single protein complex per sample. This rate is similar to what is possible using AP- and IP-MS methods.
A QUICK LC-MS Method to Identify Specifically-Bound Proteins
As mentioned previously, differentiating specific and nonspecific PPI is a major challenge in these types of studies. Optimizing the protein complex extraction conditions is critical, however, it is impossible to reflect in vivo conditions using an in vitro process.
Unfortunately, MS analysis only identifies which proteins are present, it does not indicate which are specifically bound to the target protein. Mass spectrometry signals themselves are not inherently quantitative, however, certain components of the MS data can allow some conclusions about the relative abundance of a particular protein in a sample to be made. One of these components is the number of peptides identified for a specific protein. This analysis, known as spectral counting, is based on the premise that the more abundant a protein is in a sample, the greater chance that its peptides will be identified using MS [36]. While the confidence level in spectral counting results is highest for proteins identified in experimental samples and completely absent in negative controls, there are many instances where a protein will be identified in both samples, albeit by different numbers of peptides.
Unfortunately, spectral counting results are not very reliable for proteins identified by only one or two peptides; a situation that is often observed in PPI studies. Fortunately, Matthias Mann’s laboratory developed a rapid, MS-based, strategy that utilizes stable isotope labeling to discriminate specifically and non-specifically bound members of a protein complex (Figure 3). This strategy, termed QUICK (Quantitative Immunoprecipitation Combined with Knockdown), combines Stable-Isotope Labeling with Amino Acids in Cell culture (SILAC) with IP of a target protein, RNA interference (RNAi), and MS analysis [37,38]. In the QUICK method, cells are cultured in medium containing a heavy isotope-substituted amino acid (e.g., 13C6 lysine), so that every protein contains a heavy version of that amino acid. A separate culture of the same cells is grown using identical conditions, except that the medium does not contain the heavy isotope-substituted amino acid. RNAi is added to one of the cultures to knockdown the expression of the target protein. The complex bound to the target protein is then extracted from the cultures separately using the exact same conditions. Once extracted, the two IP samples are mixed and analyzed using MS. Proteins that are non-specifically bound to the protein complex, or components of the IP process (e.g., antibody and solid support) will appear as a doublet of peaks in the resulting MS spectra with an area ratio of approximately 1:1. Proteins that are specifically-bound members of the protein complex will ideally be represented by only a single peak originating from the non-RNAi treated culture. If the RNAi treatment does not completely knockdown protein expression, specifically bound proteins will be seen as a doublet with the peptide peaks originating from the RNAi-treated culture being much less intense.
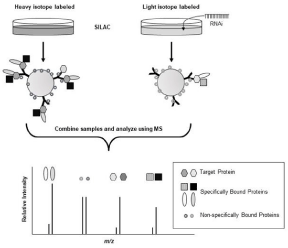
Figure 3: Principles of Quantitative Immunoprecipitation Combined with Knockdown (QUICK). Stable-Isotope Labeling with amino Acids in Cell Culture (SILAC)
is used to label proteins in samples being compared. Target protein expression is knocked down in one of the cultures using RNA interference (RNAi). Protein
complexes are separately extracted from each culture using immunoprecipitation. At this point the complexes are combined and analyzed using Mass Spectrometry
(MS). Non-specifically bound proteins are recognized by having two peaks (i.e., heavy and light isotope labeled versions) of equal intensity. Specifically-bound
proteins are represented by two peaks, with the peak originating from the culture to which RNAi was applied being of much of lower intensity than its counterpart
extracted from the untreated sample.
The QUICK method has been applied to a number of PPI studies, including one for identifying binding partners to 14-3-3ζ [39]. The MS analysis resulted in the identification of 292 proteins as part of the 14-3-3ζ complex. Fifty-one of the proteins that were quantified were identified as background based on the MS data. About 67% of the proteins identified in this study had been reported in previous studies [40]. More interestingly, 95 new 14-3-3ζ interacting partners were identified. Six proteins that had been previously reported as putative 14-3-3ζ binding partners (GAPDH, regulator of nonsense transcripts 1, fructose-bisphosphate aldolase A, L-lactate dehydrogenase A chain, L-lactate dehydrogenase B chain, and uncharacterized protein MVP) were characterized in this QUICK study as being non-specifically interacting proteins. Functional classification of the identified 14-3- 3ζ interacting proteins showed that almost one-third of the proteins were involved in metabolism. The other major represented categories including signal transduction (11%), apoptosis (10%), and nucleic acid binding (7%). Taken together, these results show that 14-3-3ζ is a critical regulator of a diverse group of biological functions. This study also demonstrated that QUICK is a useful approach to detect specific PPI with very high confidence and may have a wide range of applications in the investigation of protein complexes. The QUICK method is also amenable to analyzing protein complexes extracted using different methods and is not limited to RNAi-treated cells. While most QUICK studies have been performed using in vitro samples, it is amenable to in vivo analysis with the advent of stableisotope labeling of mammals [41].
Applications of PPI Studies
Characterization of yeast mitochondria using crosslinking mass spectrometry
A study published by Dr. Christophe Borchers, a pioneer in the application of CX-MS for identifying PPI, illustrates the power of this technique. This study extracted mitochondria from the yeast strain YPH499 [24]. To preserve PPI, the mitochondria were extracted using non-denaturing conditions and gently diluted to 5mg/ml in isotonic buffer (250mM sucrose, 1mM EDTA, 10mM MOPS-KOH, pH 7.2). Proteins within the mitochondria were cross-linked using the crosslinking reagent Cyanurbiotindipropionylsuccinimide (CBDPS) at a concentration of 2mM. To minimize artifactual cross-linking, the reaction was quenched by adding ammonium bicarbonate. After collecting the cross-linked mitochondria using centrifugation, they were immediately lysed by placing them in a hypotonic buffer followed by sonication.
After isolating the protein in pellet form, it was proteolyzed using trypsin. The tryptic peptides were washed using various solutions and preconcentrated prior to initial separation into 19 fractions using strong cation exchange chromatography. The crosslinked peptides were enriched from each of these aliquots using monomeric avidin beads as the CBDPS reagent contains a biotin group. The enriched samples were then prepared for MS analysis to identify the crosslinked peptides.
The analysis of the yeast mitochondria identified 751 unique cross-linked inter-protein pairs involving 264 yeast mitochondrial proteins, or 20% of the currently characterized yeast mitochondrial proteome. These proteins represented a total of 338 unique PPI, representing the most comprehensive set of yeast mitochondrial PPI determined as of yet using this technique. Over 70% of the identified PPI had not previously been described as part of the EMBL-EBI IntAct Molecular Interaction Database, which curates PPI reported in literature [42]. The impartiality of the linker is illustrated by the fact that soluble, peripheral, and integral protein classes accounted for 31%, 29%, and 24% of the proteins, respectively. Not only did the observed interactions make biological sense, the most observed sub-compartment localization pairs were between inner-membrane proteins (81PPIs), inner-membrane and matrix proteins (52PPIs), and matrix proteins (51PPIs). Very few non-sensical interactions, such as PPI between outer-membrane and matrix proteins, were observed.
As with any large discovery-driven effort, validation is necessary to substantiate the study results. For cross-linking studies, this validation focuses on evaluating if the peptides that were identified as being crosslinked are within the length of the cross-linking reagent. This type of validation is necessarily done using known three dimensional structures of proteins identified in the study. In this study, the results were validated by mapping the identified crosslinks to existing structural models of the electron transport chain protein complexes available in the Protein Data Bank (PDB) database [43]. The proteins that were chosen for validation included ATP synthase (6B8H), complex III2 (3CX5), complex V (6CP3), and complex V dimer (6B8H). The observed Ca-Ca distance distributions were plotted against distances of possible random crosslinks for these four complexes. The correlation between observed and possible crosslinks showed good correlation with most of the observed crosslinks being less than 38Å (i.e., the length of the CBDPS reagent).
Identification of PPI in mammalian brain using cofractionation mass spectrometry
The current state and potential of PPI studies was reflected in a recent study that generated an interaction map of various regions of the mouse brain [44]. This study utilized a CF-MS approach to separate protein complexes extracted from homogenized brains dissected from 12 week old male CD1 mice. The brain tissue was extracted using a non-denaturing lysis buffer, followed by extraction of the resulting pellet using two sequential methods; one detergent free extraction followed by subsequent extraction using a detergentcontaining buffer. The protein extracts were fractionated using various combination of Isoelectric Focusing (IEF), Ion Exchange Chromatography (IEX) and Heparin-Ion Exchange Chromatography (HIEX). The result of the various fractionation steps, along with replicate analyses to evaluate reproducibility, was the analysis of 550 fractions using MS to identify the proteins within each fraction.
This enormous study resulted in the putative identification of over 5700 proteins that were assigned within 1030 protein complexes. There were obviously many proteins that were observed in more than one complex, however, in these instances the uniqueness of each complex could be determined using subcellular compartmentalization data. For example, a number of complexes associated with axons, dendrites, and synapses were observed. Within this collection were complexes 42 and 51, which shared 14 common SNARE proteins that are necessary for synaptic-vesicle docking [45]. These two complexes were differentiated by the fact that complex 42 included synaptic-vesicle transmembrane factors (Sv2b, Slc4a10, and Prrt2), while complex 51 contained proteins that mediated ER-Golgi vesicle transport and fusion (Vcp, Sec22b, Scfd1, and Arfgap2). Complexes 234 and 267, which contained core groups of proteins involved in glutamatergic neurotransmission, possessed key differences in their other interacting proteins. For example, complex 234 contained proteins involved in synapse excitation (voltage-dependent anion channel 1, neuroligin-2, and solute carrier family 17 member 6), while complex 267 contained Ras-related protein Rab-21 and integrin subunit beta 1, two proteins involved in endosomal trafficking. These results demonstrate that although complexes may contain similar core proteins, differences in other peripheral members of the complex can alter the complex’s function.
Previously unreported PPI identified in this study are significantly enriched for proteins involved in RNA metabolism, messenger RNA processing, and binding. These assemblies typically comprise Ribosomal Binding Proteins (RBPs) involved in the biogenesis, distribution, and metabolism of coding and non-coding RNAs [46]. These complexes ranged in size from 8 (complex 250) to well over a dozen RBPs (e.g., complex 22, which contained 28 RBPs). Key discoveries within complex 22 were further evaluated. In particular, interactions involving TDP-43 (TAR DNA-binding protein 43) and HNRNP1 (heterogeneous nuclear ribonucleoprotein H) were evaluated using co-IP of either protein from brain cortices of wild-type mice. Immunoprecipitation of TDP-43 co-precipitated endogenous HNRNPH1, DDX5 (DEAD-box helicase 5) and TIA1 (cytotoxic granule associated RNA binding protein). Likewise, IP of endogenous HNRNPH1 reciprocally pulled-down TDP-43, DDX5, TIA1, and FUS (heterogeneous nuclear ribonucleoprotein P2). Of particular interest was the detection of TDP-43 as a component of complex 168, which contained a number of other proteins involved in ribosomal binding.
These two complexes (22 and 168) were of particular interest since multiple ribosomal binding proteins that are genetically linked to Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Degeneration (FTD) were found within both. Specifically, mutations in TARDBP [47], FUS/TLS [48], and TIA1 [49] lead to the accumulation of pathological insoluble cytoplasmic inclusions in motor and cortical neurons. Mutations within the gene that expresses at Ataxin-2 (ATXN2), which was found as part of complex 22, are also associated with ALS and spinal cerebellar ataxia [50,51].
In a follow up experiment demonstrating the value of the largescale PPI studies, the investigators studied an ALS mouse model in which TDP-43 was over-expressed (TDP-43WT/WT) and ATXN2 was under-expressed (ATXN2+/-) [44]. A rapid degeneration of motor neurons was observed in mice overexpressing TDP-43. Conversely, depletion of ATXN2 (a component of complex 22) in a transgenic mouse overexpressing TDP-43 (i.e., TDP-43WT/ WTATXN2+/-), reduced TDP-43 aggregation. The overall effect of ATXN2 reduction was an increase in motor neuron survival and extended lifespan.
Challenges in generating global PPI maps
Studies that attempt to generate entire PPI maps of cells are not without their challenges. The greatest challenge is the significant number of false positive and false negative results. False negative results are the hardest to recognize as it is virtually impossible to identify what is not observed in a dataset generated using a discoverydriven approach. False positive interactions cannot be absolutely eliminated by optimizing sample preparation conditions since these discovery-driven studies cannot unequivocally optimize these methods for every complex within cells. While binary validation of each interaction using physical techniques such as co-IP, immunofluorescence, etc. would be considered the gold-standard, it is impractical for any laboratory to complete this amount of work prior to releasing the entire cellular PPI. Large PPI studies rely heavily on software algorithms that provide statistical measures of the reliability of the individual protein interactions detected within the empirical datasets. Many of these algorithms rely on databases of curated literature results that have previously demonstrated a potential interaction between specific proteins. Global PPI studies also rely on other types of empirical data, such as interacting proteins having similar quantitative values, to substantiate their findings. As PPI studies continue in number, their accuracy can be enhanced by adding a spatial parameter into the experimental procedure. This parameter can be added by conducting subcellular fractionation into the procedure prior to protein complex separation and analysis.
While a subset of PPI can be validated using orthogonal methods to determine the study’s overall veracity, today’s technology affords a much more efficient strategy that allows the data to be disseminated throughout the scientific community. For example, the data obtained from the previously discussed mouse brain PPI study has been made publicly available within the Brain Interaction Map (BraInMap) [52]. BraInMap provides a clickable list of all 5,798 proteins identified in the study along with the 1030 complexes they were identified within. The complexes range from 3 to 110 proteins in size with many proteins being members of more than one complex. The complexes can be searched by individual protein, subcellular location, keyword, or disease association. The protein complexes can be displayed as connectivity diagrams with lines representing putative interactions found within the dataset. Proteins that are associated with neurological disorders are highlight as are those that make up the core of the complex.
PPI databases
Since it is practically and fiscally impossible for individual laboratories to manually validate all of the data identified in a global PPI study, databases containing PPI data have been developed and made publicly available. A brief list of some of the available PPI databases is provided in Table 2. These databases are generally classified into three categories; primary, secondary, and predictive databases. Primary databases collect experimental interaction data from peer-reviewed publications. Secondary databases collect data from several primary databases and collate them into a single, integrated data repository. Predictive databases are comprised of experimentally inferred data acquired from primary databases but also uses computational methods to predict the existence of molecular interactions. Many of the databases provide software tools that allow interactions to be visualized and queried. Ultimately, the value of these databases is not the summaries of the data they provide, but rather the opportunity for investigators to evaluate their experimental data against previous reports and generate new hypotheses.
![]()
Database
Description
URL
Database of Interacting Proteins
Catalogs experimentally determined PPI curated from literature by both expert scientists and computational approaches.
uniprot.org/database/DB-0016
Human Protein Reference Database
Contains information related to interaction networks, post-translational modifications, domain architecture, and disease association for proteins in the human proteome.
Data obtained from literature that has been manually evaluated by experts interpreting and analyzing published data.hprd.org/
IntAct
Protein interactions derived from literature curated by experts and experimental results submitted by researchers.
ebi.ac.uk/intact/
CORUM
Database of manually curated protein complexes from mammals. Data is annotated with features including complex function and subcellular localization.
mips.helmholtz-muenchen.de/corum/
The Molecular INTeraction database
Database of experimentally verified PPI curated from scientific literature by expert curators.
mint.bio.uniroma2.it/
Reactome
Database of interactions expertly curated from primary literature. Provides software tools to support data visualization, integration and analysis across many different types of biomolecules.
https://reactome.org/
BIOPLEX
Experimental PPI data obtained using affinity purification-mass spectrometry analysis of 293T and HCT116 cell lines.
wren.hms.harvard.edu/bioplex/
Table 2: List of available websites for studying Protein-Protein Interactions (PPI).
Conclusion
Technology developments made in the past couple of decades have brought tremendous advances in how, and how fast, protein complexes are identified. While this review describes some of the predominant methods, even it is incomplete as improvements for characterizing PPI are regularly being generated. The biggest improvement over the past couple of decades has been the strategy for identifying PPI. Basic research used to be limited to verifying or disproving hypothetical interactions; however, methods for routinely discovering novel interactions using discovery-driven techniques are now common. There are, however, many challenges that advanced discovery-driven technologies cannot yet overcome. Limiting the number of non-specifically bound proteins identified within these complexes will be central for generating PPI maps that truly reflect cell physiology. While the lack of similarity between some of the earliest attempts at generating cell-wide PPI maps was initially disappointing [8,9], these studies generated the motivation for other groups to investigate methods to improve these types of analyses. Whether repeat analyses or continued comparison amongst large interactome datasets will provide an accurate view of the protein circuitry within the cell remains to be seen. Presently, these discoverybased methods for identifying PPI provide “possibilities” that must be confirmed before any certain biological function can be established. Finally, publicly available databases will be crucial as they provide informational repositories that can be scrutinized and queried by multitudes of researchers with the common goal of generating an accurate model of PPI within cells and living organisms.
Acknowledgment
The author would like to thank Cedarville University for support during the writing of this manuscript.
References
- Seychell BC, Beck T. Molecular basis for protein-protein interactions. Beilstein J Org Chem. 2021; 17: 1-10.
- Rossi MJ, Mazin AV. Rad51 protein stimulates the branch migration activity of Rad54 protein. J Biol Chem. 2008; 283: 24698-24706.
- Zofall M, Grewal SI. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol Cell. 2006; 22: 681-692.
- Hara T, Abe M, Inoue H, Yu LR, Veenstra TD, Kang YH, et al. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr-341 in G2/M phase. Oncogene. 2006; 25: 566-578.
- Zhou M, Veenstra TD. Proteomic analysis of protein complexes. Proteomics. 2007; 7: 2688-2697.
- Shiio Y, Itoh M and Inoue J. Epitope tagging. Methods Enzymol. 1995; 254: 497-502.
- Goto-Silva L, Maliga Z, Slabicki M, Murillo JR, Junqueira M. Application of shotgun proteomics for discovery-driven protein-protein interaction. Methods Mol Biol. 2014; 1156: 265-278.
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006; 440: 637-643.
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006; 440: 631-636.
- Tuite MF. Strategies for the genetic manipulation of Saccharomyces cerevisiae. Crit Rev Biotechnol. 1992; 12: 157-188.
- Goll J, Uetz P. The elusive yeast interactome. Genome Biol. 2006; 7: 223- 228.
- Yates JR 3rd. Mass spectrometry. From genomics to proteomics. Trends Genet. 2000; 16: 5-8.
- Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000; 405: 837-846.
- Liu Y, Trnka MJ, Guan S, Kwon D, Kim DH, Chen JJ, et al. A novel mechanism for NF-κB-activation via IκB-aggregation: Implication for hepatic Mallory- Denk-Body induced inflammation. Mol Cell Proteomics. 2020; 19: 1968-1986.
- Pu S, Vlasblom J, Turinsky A, Marcon E, Phanse S, Trimble SS, et al. Extracting high confidence protein interactions from affinity purification data: at the crossroads. J Proteomics. 2015; 118: 63-80.
- Siva Sankar D, Dengjel J. Protein complexes and neighborhoods drive autophagy. Autophagy. 2020; 13: 1-17.
- Wong KS, Snider JD, Graham C, Greenblatt JF, Emili A, Babu M, et al. The MoxR ATPase RavA and its cofactor ViaA interact with the NADH: ubiquinone oxidoreductase I in Escherichia coli. PLoS One. 2014; 9: e85529.
- Fujii T, Yamasaki R, Kira JI. Novel neuropathic pain mechanisms associated with allergic inflammation. Front Neurol. 2019; 10: 1337-1344.
- Park DI, Turck CW. Interactome studies of psychiatric disorders. Adv Exp Med Biol. 2019; 1118: 163-173.
- Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. CK2 is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007; 17: 179-184.
- Pang CNI, Ballouz S, Weissberger D, Thibaut LM, Hamey JJ, Gillis J, et al. Analytical guidelines for co-fractionation mass spectrometry obtained through global profiling of gold standard Saccharomyces cerevisiae protein complexes. Mol Cell Proteomics. 2020; 19: 1876-1895.
- McWhite CD, Papoulas O, Drew K, Cox RM, June V, Dong OX, et al. A panplant protein complex map reveals deep conservation and novel assemblies. Cell. 2020; 181: 460-474.
- Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, et al. Panorama of ancient metazoan macromolecular complexes. Nature. 2015; 525: 339-344.
- Makepeace KAT, Mohammed Y, Rudashevskaya EL, Petrotchenko EV, Vögtle FN, Meisinger C, et al. Improving identification of in-organello proteinprotein interactions using an affinity-enrichable, isotopically coded, and mass spectrometry-cleavable chemical crosslinker. Mol Cell Proteomics. 2020; 19: 624-639.
- Petrotchenko EV, Borchers CH. Crosslinking combined with mass spectrometry for structural proteomics. Mass Spectrom Rev. 2010; 29: 862- 876.
- Rey M, Dhenin J, Kong Y, Nouchikian L, Filella I, Duchateau M, et al. Advanced in vivo cross-linking mass spectrometry platform to characterize proteome-wide interactions. Anal Chem. 2021; 93: 4166-4174.
- Basu A, Ash PE, Wolozin B, Emili A. Protein interaction network biology in neuroscience. Proteomics. 2021; 21: e1900311.
- Qin W, Cho KF, Cavanagh PE, Ting AY. Deciphering molecular interactions by proximity labeling. Nat Methods. 2021; 18: 133-143.
- Habel JE. Biotin proximity labeling for protein-protein interaction discovery: The BioID method. Methods Mol Biol. 2021; 2261: 357-379.
- Frankenfield AM, Fernandopulle MS, Hasan S, Ward ME, Hao L. Development and comparative evaluation of endolysosomal proximity labeling-based proteomic methods in human iPSC-derived neurons. Anal Chem. 2020; 92: 15437-15444.
- Arora D, Abel NB, Liu C, Van Damme P, Yperman K, Eeckhout D, et al. Establishment of proximity-dependent biotinylation approaches in different plant model systems. Plant Cell. 2020; 32: 3388-3407.
- Udeshi ND, Pedram K, Svinkina T, Fereshetian S, Myers SA, Aygun O, et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat Methods. 2017; 14: 1167-1170.
- Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016; 353: 1123-1129.
- Li J, Han S, Li H, Udeshi ND, Svinkina T, Mani DR, et al. Cell-surface proteomic profiling in the fly brain uncovers wiring regulators. Cell. 2020; 180: 373-386.e15.
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018; 36: 880-887.
- Carvalho PC, Hewel J, Barbosa VC, Yates JR 3rd. Identifying differences in protein expression levels by spectral counting and feature selection. Genet Mol Res. 2008; 7: 342-356.
- Selbach M, Mann M. Protein Interaction Screening by Quantitative Immunoprecipitation Combined with Knockdown (QUICK). Nat Methods. 2006; 3: 981-983.
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002; 1: 376-386.
- Ge F, Li WL, Bi LJ, Tao SC, Zhang ZP, Zhang XE. Identification of novel 14- 3-3zeta interacting proteins by Quantitative Immunoprecipitation Combined with Knockdown (QUICK). J Proteome Res. 2010; 9: 5848-5858.
- Stelzer G, Rosen R, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards suite: From gene data mining to disease genome sequence analysis. Curr Prot Bioinformatics. 2016; 54: 1.30.1-1.30.33.
- McClatchy DB, Yates JR 3rd. Stable Isotope Labeling in Mammals (SILAM). Methods Mol Biol. 2014; 1156: 133-146.
- Hermjakob H, Luisa Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, Orchard S, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004; 32: D452-455.
- Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Di Costanzo L, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019; 47: D464-D474.
- Pourhaghighi R, Ash PEA, Phanse S, Goebels F, Hu LZM, Chen S, et al. BraInMap elucidates the macromolecular connectivity landscape of mammalian brain. Cell Syst. 2020; 10: 333-350.
- Chen X, Tomchick DR, Kovrigin E, Araç D, Machius M, Südhof TC, et al. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002; 33: 397-409.
- Shi Z, Barna M. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol. 2015; 31: 31-54.
- Bigio EH. TDP-43 variants of frontotemporal lobar degeneration. J Mol Neurosci. 2011; 45: 390-401.
- Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res. 2010; 66: 131-133.
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, et al. TIA1 mutation in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017; 95: 808-816.e9.
- Becker LA, Gitler AD. Ataxin-2 is droppin’ some knowledge. Neuron. 2018; 98: 673-675.
- Dansithong W, Paul S, Figueroa KP, Rinehart MD, Wiest S, Pflieger LT, et al. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 2015; 11: e1005182.
- Pourhaghighi. Macromolecular connectivity landscape of mammalian brain.