
Research Article
Austin Biomark Diagn. 2014;1(1): 4.
QX-314 Induces Analgesia to Nociceptive Thermal Stimulus by Co-Application with Capsiate or Anandamide
Nakagawa H1* and Hiura A2
1Pediatric Dentistry, Tokushima University Hospital, Japan
2Department of Oral Histology, University of Tokushima, Japan
*Corresponding author: Nakagawa H, Pediatric Dentistry, Tokushima University Hospital, Tokushima, 3-18-15 Kuramoto cho, Tokushima 770-8054, Japan.
Received: Sep 12, 2014; Accepted: Oct 13, 2014; Published: Oct 14, 2014
Abstract
Recently, N-ethyl-lidocaine (QX-314) was demonstrated to pass through a nociceptor membrane when the drug was co-applied with capsaicin, thereby leading to analgesia due to selective blockade of a sodium channel associated with the excitability in nociceptors. However, capsaicin induces a pain in the case of injection with QX-314. Therefore this good idea is unfavorable for a clinical therapy. Then, we exam–ined whether non pungent Capsiate (CST) and arachidonylethanolamide (anandamide: AEA) can deliver QX-314 into nociceptive neurons through the pores of Transient Receptor Protein Vanilloid 1 (TRPV1) and contribute to the antinociceptive effect. Dimethyl sulfoxide (DMSO), CST, QX-314, a mixture (combination) of CST and QX-314 (CST/ QX-314), and a combination of AEA and QX-314 (AEA/QX-314) were injected into the hind paws of rats to evaluate anti-noxious heat effect. A long-lasting sensory nerve block (analgesia) was induced by CST/QX-314 or AEA/QX-314. Especially, the CST/QX-314 caused more effective than our previous studies with the combination of capsaicin and QX-314 (CAP/QX-314). The present results also indicate that CST can induce analgesia by itself. The study aimed at developing a new method with substances distinct from capsai–cin to eliminate the action-potential firing in nociceptors (but effective only for pain sensation). As a result, CST is likely to be a potent candidate for pain relief.
Keywords: Anesthetics; Capsiate; Anandamide; QX-314; Noxious heat tests
Introduction
Local anesthetic can produce reversible suppression of the nerve conduction when applied to the peripheral nociceptors. The drugs block the voltage-gated sodium channel, leading to loss of pain or hypoalgesia in the applied area [1]. An anesthetic lidocaine, well used clinically, is a tertiary amine that possesses a mixed state of prorogated and uncharged base forms under physiological conditions [2]. Because the uncharged hydrophobic form of lidocaine can penetrate all neuronal membranes, it affects on multiple functions other than anesthesia; in addition to numbness by blockade of the low threshold sensory nerve, dyskinesia and insufficient physical conditions occur due to the blockades of motor nerve and the autonomic nerve, respectively.
Binshtok et al [3] devised a new method for pain relief using permanently charged sodium channel blockers such as N-ethyllidocaine (QX-314), a lipophobic lidocaine derivative. QX-314 can block sodium channel when injected directly into the cytoplasm of the cell. On the contrary, when QX-314 was applied extra cellular in a standard local anesthetic, it cannot cut off a sodium channel because of failure to penetrate the membrane by its lipophobic nature [4,5]. However, QX-314is able to selectively enter into nociceptors if coapplied with capsaicin and lead to a preferential blockade of sodium channel responsible for the excitability on nociceptors [3,6]. The pore size of the Transient Receptor Protein Vanilloid 1 (TRPV1) channels, which are opened by capsaicin, is enough to deliver QX-314 into nociceptive sensory neurons [3,6]. Quite recently, the passage of QX- 314 through TRPV1 channel was visually confirmed by use of [5,6] – Carboxyl fluoresce in-conjugated QX-314 [7]. Therefore, the new method without numbness and motor paralysis was expected for a selective blockade of nociception. Since capsaicin produces a pain when it is co-applied with QX-314, this improvable method cannot be recommended for a clinical use. As lidocaine can activate TRPV1 channel, QX-314 was assumed to enter into a nociceptor through TRPV1 channel by co-application with lidocaine [8,9].
Unexpectedly co-application of QX-314 and lidocainecuts off the excitability of various types of neurons other than nociceptors [8,9]. The purpose of this study is to find the material in place of capsaicin to develop a new method in the clinical setting. In the previous studies, we examined the ability of allylisothiocyanate (AITC) and menthol for delivery of QX-314 into nociceptive neurons through the pores of both Transient Receptor Potential Ankyrin 1 (TRPA1) and transient receptor potential melastatin-8 (TRPM8), respectively, and whether they can produce antinociceptive effects or not [7]. The results indicated that AITC (via TRPA1 channel) and melastatin-8 (via TRPM8 channel) are ineffective in the transport of QX-314 compared with capsaicin (via TRPV1 channel) [7]. Therefore, it is meaningful to search a new TRPV1 agonist with no irritation like capsaicin.
For the first time, capsiate (CST) and arachidonylethanolamide (AEA: an andamide) were chosen as candidates. CST is extracted from the fruit of non-pungent cultivar of the pepper (CH-19 Sweet species), and its chemical structure is very similar to capsaicin except the ester bond in substitution for an amide bond between vanillyl structure and fatty acid chains (Figure 1) [10,11]. Therefore, it is called capsinoid different from capsaicinoid as an analog of capsaicin. Iida et al. reported that CST with high lipophilicity is an agonist of TRPV1, exciting peripheral nociceptors when subcutaneously injected into hind paws but not skin of mice [12]. About 20 years ago, AEA was found as a component binding to the cannabinoid receptor in the brain (Figure1)? Continuously, multiple new receptors for AEA were identified such as TRPV1, and demonstrated that AEA has an ability to activate TRPV1 [13]. At present, the AEA is often introduced as “endovanilloid” [13]. The ability of CST and AEA to deliver QX-314 into nociceptive neurons via theTRPV1channel and whether they can produce an antinociceptive effect were investigated in this study.
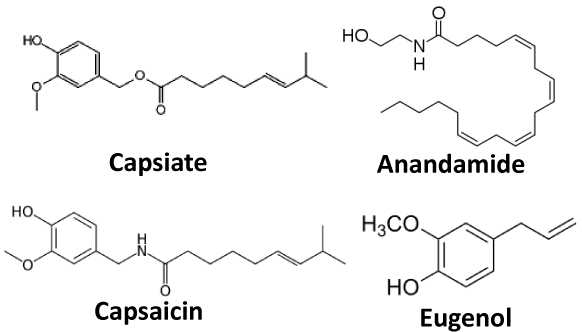
Figure 1: Chemical structure of capsiate, anandamide, capsaicin, and eugenol Capsiate, called capcinoid, is an analog of capsaicin. Anandamide and eugenol have a similar structure (vanillyl moiety) to capsaicin, suggesting actions onTRPV1channel.
Materials and Methods
Animals
Thirty-six male Wister rats (120–350 g) were used for behavioral tests. The animals were housed in a controlled lighting environment (12h in light and 12h in dark cycle) with rodent feed and water available ad libitum. Behavioral tests started at the same time in a day to avoid circadian effects. The study protocol was approved by the Tokushima University Care and Use of Animals Committee.
Application of chemicals
Drugs: Capsiate (CST) was presented by Ajinomoto Co., Ltd (Japan). Anandamide (AEA) and QX-314 were purchased from Cayman Chemical (USA) and Enzo Life Science (Japan), respectively. CST and AEA were freshly prepared with dimethyl sulfoxide (DMSO). QX-314 was dissolved in physiological saline.
Intraplantar injection with agonists for behavioral tests: Ten μL (in each treatment) of 25mM CST (CST group), 2% QX-314 only (QX-314 group), a mixture of CST and QX-314 (CST/QX-314 group), and DMSO alone (DMSO group) were injected into the rat right plantar hind paws of rats (CST experiments). Similarly, 10μL of 5mM AEA were injected to evaluate the effects of AEA (AEA experiments). Six animals were used in each experimental group. The data from the CST and AEA experiments were compared with those of QX-314 and DMSO groups. The concentration of the drugs was determined according to the references [12,13].
Hind paw withdrawal behavioral tests to noxious heat (Hargreaves method): Thermal sensitivity was examined by exposing the hind paws to defined radiant heat stimuli through a transparent glass surface (Plantar Test; Ugo Basile Srl, Comerio, Italy). The paw-withdrawal latencies were started to record just after the animals were put on the transparent glass [14]. The intensity of the heat stimuli was adjusted to 50 [15]. A cut-off time was set at 20 seconds to avoid the tissue damage. Each rat received two serial stimuli at intervals of at least 2 minutes. Before the drug injection, baseline withdrawal response latencies were determined for all of the animals. The withdrawal-response latencies were measured at 10, 30, 60, 120, 180, 240, and 300 minutes after the drug injections. The experimenter was blind to the treatment groups in all behavioral tests.
Data analysis
The experimental data were analyzed with repeated measures analysis of variance (ANOVA), followed by the Least Significant Difference (LSD) post hoc test. Results were expressed as the mean ± Standard Error (SE). P <0.05 was considered as significant differences.
Results and Discussion
CST/QX-314 induced longer withdrawal latency (analgesia) than either the QX-314 or DMSO between 10 and 300 minutes after injection (P < 0.001, Figure 2). Although CST alone induced analgesia between 10 and 60 minutes after injection (10 min: P < 0.01, 30 – 60 min: P < 0.001, Figure 2), the CST/QX-314 elicited a more intense analgesia than the CST between 10 and 300 minutes except for 30 minutes after injection (P < 0.001). AEA/QX-314 also induced a longer withdrawal latency between 60 and 240 minutes after injection (60 min: P < 0.01, 120 – 240 min: P < 0.001, Figure 3).Thus, alonglasting sensory nerve inhibition was demonstrated in both CST/ QX-314 and AEA/QX-314 treatments. Even the analgesia by CST alone was also sustained for longer duration than AEA. The maximal thermal latency was 7.9 ± 1.7 seconds in the CST/QX-314, whereas that was 5.8 ± 1.1 seconds in the AEA/QX-314; the analgesic potency of CST was significantly 1.4 times higher than the AEA. The analgesic potency of CST was high at approximately 1.2 times compared with capsaicin (CAP/QX-314) from our previous data [7], but it was not significant. Taken together, these drugs (CST, AEA and CAP) have a common duration of analgesia which lasted between 60 and 240 minutes after treatment. The appearance of maximal analgesia also coincides with between 120 and 180 minutes in each drug. Probably, some definite time is a necessity to enter into nociceptors and followed by inducing analgesia.
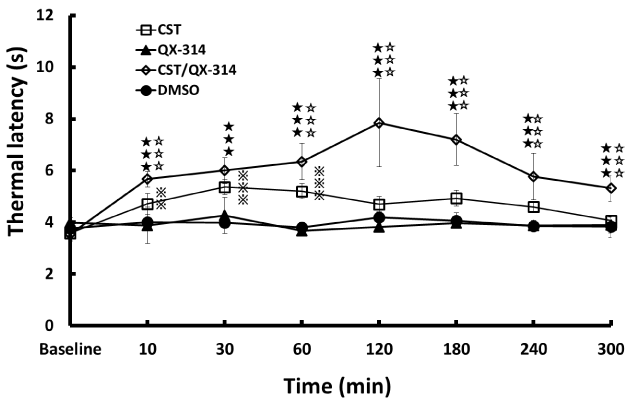
Figure 2: Effects on noxious thermal sensitivity after co-application of CST and QX-314 CST/QX-314 significantly caused longer withdrawal latency (analgesia) than either the QX-314 or DMSO between 10 and 300 minutes after treatment (P < 0.001). The CST/QX-314 induced more intense analgesia than the CST between 10 and 300 minutes (but except for 30 minutes) after treatment (P < 0.001). The CST alone also induced analgesia between 10 and 60 minutes after treatment (10 min: P < 0.01, 30 – 60 min: P < 0.001).♦♦♦: p<0.001 (CST/QX-314 vs. QX-314 or DMSO), ◊◊◊: p<0.001 (CST/QX-314 vs. CST, ℑℑℑ: p<0.001, ℑℑ: p<0.01 (CST vs. QX-314 or DMSO).
Figure 3: Effect on noxious thermal sensitivity after co-application of AEA and QX-314 AEA/QX-314 significantly induced longer withdrawal latency (analgesia) between 60 and 240 minutes after treatment (60 min: P < 0.01, 120 – 240 min: P < 0.001). Note that the response behaviors (curves) are very similar to that of CST/QX-314. ♦♦: p<0.01, ♦♦♦: p<0.001(AEA/QX-314 vs. AEA, QX-314 or DMSO).
As mentioned above, the effects (analgesia) of CST/QX-314 appeared rapidly and lasted longer than CAP/QX-314 and AEA/QX- 314. CST is reported to potentiate energy metabolism by stimulating the sympathetic nervous system like capsaicin [16]. Because CST is able to activate TRPV1 and open the channel similarly to capsaicin [12], the QX-314 could penetrate via TRPV1 channel when it is coadministered with CST. The CST also caused more potent analgesia between 10and 60 minutes after injection, suggesting that the CST elicited a local analgesia by itself. Eugenol (A dental analgesic extracted from clove oil), is frequently used in dental treatment to recover toothache. It is a small molecule with structural similarity to capsaicin, thereby displaying an agonist function for the TRPV1 receptor (Figure 1). Curiously, eugenol inhibited Na+ channels in a TRPV1-independent manner [17-19]. The CST is assumed to restrain Na+ channels not via TRPV1 likely to eugenol. Tomohiro et al. [20] examined the influences of CST on the capsaicin action potential by applying the air-gap method to the frog sciatic nerve [20]. They demonstrated the action potential inhibition by CST in a wide range of concentration [20]. Furthermore, Shintaku et al. [21] reported that CST could activate TRPA1 using Ca2+- imaging and whole-cell patchclamp methods. High concentration (2mM) of TRPA1 agonists (AITC) had an ability to inhibit nerve conduction regardless of the TRPA1channel [22]. Thus, high concentration of CST (25mM) may induce anesthetic efficiency as well.
Since the discovery of the cannabinoids receptor, the many studies have been undertaken to detect endogenous legends. AEA was an initial legends identified in the brain of the pig [23]. Capsaicin-related chemical formula of AEA suggests an activation of TRPV1, indicating the interaction between cannabinoids and vanilloid systems [24-26]. Accordingly, the long duration of withdrawal latency by the AEA/ QX-314 than either the QX-314 or DMSOis thought to be occurred. QX-314 appeared to enter the nociceptive sensory nerves via TRPV1 channel due to opening the channel by AEA. However, the duration of analgesia induced by AEA/QX-314 was shorter than our previous study by CAP/QX-314 [7]. The AEA/QX-314 elicited analgesia between 60 and 240 minutes. On the other hand, the CAP/QX-314 elicited analgesia between 60 and 300 minutes [7]. The binding affinity of AEA for TRPV1 seems to be weaker than that of capsaicin. In fact, Toth et al. reported that the affinity of AEA was five times weaker than capsaicin [27]. Ross also reported that the binding potency of AEA on TRPV1 is significantly lower than that of capsaicin (0.5-10 μM and 10-100 nM, respectively) [28].
Conclusion
The study showed that a long-lasting sensory nerve block was induced by injections with CST/QX-314 and AEA/QX-314. Especially, the CST/QX-314 was more effective than CAP/QX-314 in our previous studies. The results also indicated that CST alone can induce analgesia by itself. We aimed at developing a new method by finding some substances different from capsaicin to eliminate the action-potential firing in nociceptors. In this regard, CST may be a higher beneficial candidate.
Acknowledgment
We appreciate the generous offer of CPA by Ajinomoto Co., Ltd. (Japan) for this study. This work was supported by the Japan Society for the Promotion of Science; KAKENHI, grant number 22592289.
References
- Butterworth J, Oxford GS. Local anesthetics: a new hydrophilic pathway for the drug-receptor reaction. Anesthesiology. 2009; 111: 12-14.
- Butterworth JF, Strichartz GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990; 72: 711-734.
- Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007; 449: 607-610.
- Omana-Zapata I, Khabbaz MA, Hunter JC, Bley KR. QX-314 inhibits ectopic nerve activity associated with neuropathic pain. Brain Res. 1997; 771: 228-237.
- Ries CR, Pillai R, Chung CC, Wang JT, MacLeod BA, Schwarz SK. QX-314 produces long-lasting local anesthesia modulated by transient receptor potential vanilloid receptors in mice. Anesthesiology. 2009; 111: 122-126.
- Kim HY, Kim K, Li HY, Chung G, Park CK, Kim JS, et al. Selectively targeting pain in the trigeminal system. Pain. 2010; 150: 29-40.
- Nakagawa H, Hiura A. Comparison of the transport of QX-314 through TRPA1, TRPM8, and TRPV1 channels. J Pain Res. 2013; 6: 223-230.
- Binshtok AM, Gerner P, Oh SB, Puopolo M, Suzuki S, Roberson DP, et al. Co application of lidocaine and the permanently charged sodium channel blocker QX-314 produces a long-lasting nociceptive blockade in rodents. Anesthesiology. 2009; 111: 127-137.
- Roberson DP, Binshtok AM, Blasl F, Bean BP, Woolf CJ. Targeting of sodium channel blockers into nociceptors to produce long-duration analgesia: a systematic study and review. Br J Pharmacol. 2011; 164: 48-58.
- Kobata K, Sutoh K, Todo T, Yazawa S, Iwai K, Watanabe T. Nordihydrocapsiate, a new capsinoid from the fruits of a nonpungent pepper, capsicum annuum J Nat Prod. 1999; 62: 335-336.
- Luo XJ, Peng J, Li YJ. Recent advances in the study on capsaicinoids and capsinoids. Eur J Pharmacol. 2011; 650: 1-7.
- Iida T, Moriyama T, Kobata K, Morita A, Murayama N, Hashizume S, et al. TRPV1 activation and induction of nociceptive response by a non-pungent capsaicin-like compound, capsiate. Neuropharmacology. 2003; 44: 958-967.
- Toth A, Blumberg PM, Boczan J. Anandamide and the vanilloid receptor (TRPV1). Vitam Horm. 2009; 81: 389-419.
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988; 32: 77-88.
- Hiura A, Nakagawa H, Koshigae Y, Yoshizako A, Kubo Y, Ishizuka H. Age-related changes in the response to thermal noxious heat and reduction of C-fibers by neonatal treatment with capsaicin. Somatosens Mot Res. 1999; 16: 115-121.
- Ohnuki K, Haramizu S, Oki K, Watanabe T, Yazawa S, Fushiki T. Administration of capsiate, a non-pungent capsaicin analog, promotes energy metabolism and suppresses body fat accumulation in mice. Biosci Biotechnol Biochem. 2001; 65: 2735-2740.
- Cho JS, Kim TH, Lim JM, Song JH. Effects of eugenol on Na+ currents in rat dorsal root ganglion neurons. Brain Res. 2008; 1243: 53-62.
- Li HY, Park CK, Jung SJ, Choi SY, Lee SJ, Park K, et al. Eugenol inhibits K+ currents in trigeminal ganglion neurons. J Dent Res. 2007; 86: 898-902.
- Park CK, Li HY, Yeon KY, Jung SJ, Choi SY, Lee SJ, et al. Eugenol inhibits sodium currents in dental afferent neurons. J Dent Res. 2006; 85: 900-904.
- Tomohiro D, Mizuta K, Fujita T, Nishikubo Y, Kumamoto E. Inhibition by capsaicin and its related vanilloids of compound action potentials in frog sciatic nerves. Life Sci. 2013; 92: 368-378.
- Shintaku K, Uchida K, Suzuki Y, Zhou Y, Fushiki T, Watanabe T, et al. Activation of transient receptor potential A1 by a non-pungent capsaicin-like compound, capsiate. Br J Pharmacol. 2012; 165: 1476-1486.
- Matsushita A, Ohtsubo S, Fujita T, Kumamoto E. Inhibition by TRPA1 agonists of compound action potentials in the frog sciatic nerve. Biochem Biophys Res Commun. 2013; 434: 179-184.
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992; 258: 1946-1949.
- Beltramo M, Piomelli D. Anandamide transport inhibition by the vanilloid agonist olvanil. Eur J Pharmacol. 1999; 364: 75-78.
- Di Marzo V, Bisogno T, Melck D, Ross R, Brockie H, Stevenson L, et al. Interactions between synthetic vanilloids and the endogenous cannabinoid system. FEBS Lett. 1998; 436: 449-454.
- Szolcsanyi J, Jancso-Gabor A. Sensory affects of capsaicin congeners I. Relationship between chemical structure and pain-producing potency of pungent agents. Arzneimittelforschung. 1975; 25: 1877-1881.
- Toth A, Kedei N, Wang Y, Blumberg PM. Arachidonyl dopamine as a ligand for the vanilloid receptor VR1 of the rat. Life Sci. 2003; 73: 487-498.
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003; 140: 790-801.