
Review Article
Austin J Bioorg & Org Chem. 2014;1(1): 7.
Diversity-Oriented Synthesis of a Library of Podocarpic Acid Derivatives
McKee NA, Bradford S, Parish DE, Neely WC and Parish EJ*
Department of Chemistry and Biochemistry, Auburn University, USA
*Corresponding author: Parish EJ, Department of Chemistry and Biochemistry, Auburn University, Auburn, Alabama, 36849, USA.
Received: July 09, 2014; Accepted: July 22, 2014; Published: July 29, 2014
Abstract
A variety of compounds, with a related structure to podocarpic acid, have been found to possess a wide variety of biological activities, including antileukemic activity, inhibition of plant cell growth, insect toxicity and antifungal properties. In the present study, a series of synthetic derivatives of podocarpic acid have been prepared by chemical synthesis for potential biological evaluation as useful compounds for use in medicine and agriculture.
Keywords: Podocarpic acid; Derivatives; Chemical synthesis
Introduction
Podocarpic acid 1 (Figure 1) was first isolated from the resin of Podocarpus cupressins, an important timber tree which is endemic to Java, and later from Podocarpus dacrydioides ("Kahikatea") and Dacrydium cupressinum ("Rimu"), trees which are found in the timber regions of New Zealand [1]. Since 1968, more than forty oxygenated metabolites of podocarpic acid have been isolated from various species of Podocarpus [2,3].
Interest in these naturally occurring and synthetic lactones, podolactones, and related podocarpic acid derivatives has been mainly due to the novel structures of these compounds and the various types of biological activity possessed by them.
Octahydrophenanthrene lactones 2 (Figure 1) and related podocarpic acid derivatives 3 have been reported to possess hormonal and anti-inflammatory properties [4]. Other similar podolactones have been shown to inhibit the expansion and division of plant cells 4 [5-9], to have antileukemic activity 5 [10], to have antibacterial activity [11], to have insect toxicity properties [12-14], and to exhibit antitumor activity [15-18].
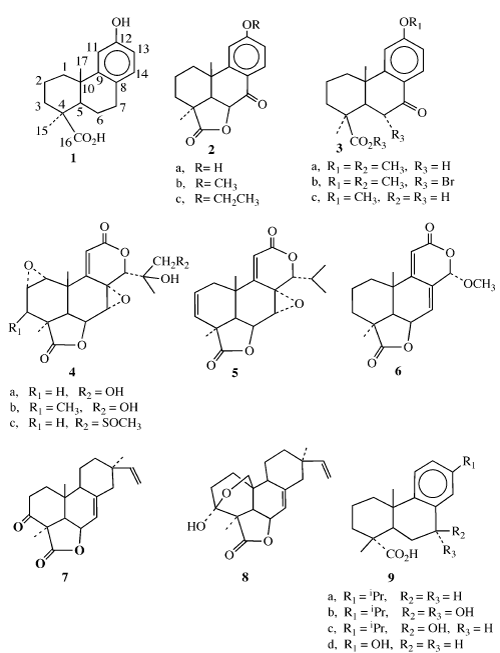
Figure 1
Other reports have indicated that these types of compounds, as a class, possess significant antifungal properties. The lactone 6 (Figure1), first isolated as a mold metabolite, was found to have significant activity against a number of fungi [19]. The momilactones A 7 and B 8 have been shown to be fungitoxic towards Cladosporium cucumerinum [20,21]. In a recent report several oxidized resin acid derivatives of dehydroabietic acid 9 (a-c) and 13-hydroxypodocarpic acid 9 (d) were found to be highly fungistatic against Dothistroma pini, a conifer pathogenic fungi [22]. It was observed that mature trees were more resistant to fungal infection and contained a greater quantity of oxidized resin acid derivatives in their resin suggesting greater resistance.
More recently, some podocarpic acid analogs have been reported as cytokine release inhibitors and this has consequently led to the discovery of novel anti-inflammatory drugs [23]. Other compounds, including a podocarpic acid anhydride, have been shown to be liver X receptor α and β agonists. These two nuclear oxysterol receptors in the liver are involved in cholesterol and lipid metabolism. Thus, any of their agonists could be useful in the development of drugs for the treatment of atherosclerosis [24].
In view of their documented biological properties, it appeared worthwhile to prepare a series of synthetic intermediates derived from podocarpic acid for potential biological activity. This report describes the preparation and characterization of these derivatives.
Results and Discussion
Commercial podocarpic acid is derived from natural sources. Several recent studies have been directed towards the total synthesis of this resin acid to assure adequate future supplies of this material for use in agriculture and medicine [25,26].
The goal of the present study was to prepare a series of derivatives related to podocarpic acid for use in structure/activity studies designed to reveal functional groups responsible for the molecules fungistatic properties. Four specific modifications were planned:
- Substitution of electron-withdrawing groups onto C (13) of the aromatic C ring (Scheme 1).
- Introduction of different halogens at C (6) (Scheme 2).
- Formation of the lactones from each 6 α -bromo methyl ester derivative (Scheme 3).
- Substitution of the methyl ester group at C (16) for an acetoxymethyl group (Scheme 4).
- Sherwood IR, Short WF. Podocarpic acid. Part I. J. Chem. Soc. 1938; 1006-1013.
- Hayashi Y, Matsumoto T. Reaction and interconversion of norditerpenoid dilactones, biologically active principles isolated from Podocarpus plants. J. Org. Chem. 1982; 47: 3421-3429.
- Cassady JM, Lightner TK, McCloud TG, Hembree JA, Byrn SR, Chang C. Revised structure of podolactone C, the antileukemic component of Podocarpus milanjianus Rendle. J. Org. Chem. 1984; 49: 942-945.
- Bible Jr RH, Grove M. Lactones of 1,12-dimethyl-6,10-dihydroxy-9-oxo-1,2,3,4,9,10,11,12-octahydrophenanthrene-1-carboxylic acid. U.S. Patent 2,753,357. 1956.
- Galbraith MN, Horn DHS, Sasse JM, Adamson D. The structures of podolactones A and B, inhibitors of expansion and division of plant cells. J. Chem. Soc. D. 1970; 170-171.
- Hayashi Y, Yokoi J, Watanabe Y, Sakan T, Masuda Y, Yamamoto R. Structures of Nagilactone E and F, and biological activity of Nagilactones as plant growth regulator. Chem. Lett. 1972; 759-762.
- Hayashi Y, Sakan T, editors. Nagilactones, plant growth regulators with an antiauxin-like activity. Proceedings of the 8th International Conference. Tokyo, Japan. 1974.
- Sasse JM, Galbraith MN, Horn DHS, Adamson DA, editors. Chemistry and biological action of podolactones and other inhibitors of plant growth. Proceedings of the 7th International Conference; 1970; Canberra, Australia. New York: Springer. 1972.
- Galbraith MN, Horn DHS, Sasse JM. Podolactones C and D, terpene sulphoxides from Podocarpus neriifolius. J. Chem. Soc. D 1971; 1362-1363.
- Bryan RF, Smith PM. An X-ray determination of the structure of podolide, an antileukemic norditerpene dilactone. J. Chem. Soc., Perkin Trans 2. 1975; 1482-1486.
- Saeki I, Sumimoto M, Kondo T. The Termiticidal substances from the wood of Podocarpus macrophyllus D. Don. Holzforschung. 1970; 24: 83-86.
- Russell GB, Fenemore PG, Singh P. Structure of hallactones A and B, insect toxins from Podocarpus hallii. Chem. Comm. 1973; 166-167.
- Singh P, Fenemore PG, Russell GB. Insect-control chemicals from plants. II. Effects of five natural norditerpene dilactones on the development of the housefly. Aust J Biol Sci. 1973; 26: 911-915.
- Singh P, Russell GB, Hayashi Y, Gallagher RT, Fredericksen S. The insecticidal activity of some norditerpene dilactones. Entomol. Exp. Appl. 1979; 25: 121-127.
- Parish EJ, Miles DH. Investigation of the antitumor activity of podocarpic acid derivatives. J Pharm Sci. 1984; 73: 694-696.
- Hayashi Y, Matsumoto T, Tashiro T. Antitumor activity of norditerpenoid dilactones in Podocarpus plants: structure-activity relationship on in vitro cytotoxicity against Yoshida sarcoma. GANN Jap. J. Canc. Res. 1979; 70: 365-369.
- Hayashi Y, Sakan T. Antitumor activity of nagilactones. Gann. 1975; 66: 587-588.
- Kupchan SM, Baxter RL, Ziegler MF, Smith PM, Bryan RF. Podolide, a new antileukemic norditerpene dilactone from Podocarpus gracilior. Experientia. 1975; 31: 137-138.
- Ellestad GA, Evans RH Jr, Kunstmann MP. Structure of a C17 antifungal terpenoid fr an unidentified Acrostalagmus species. J Am Chem Soc. 1969; 91: 2134-2136.
- Cartwright DW, Russell GE. Possible involvement of phytoalexins in durable resistance of winter wheat to yellow rust. Trans. Br. Mycol. Soc. 1981; 76: 323-325.
- Fuchs A, Davidse LC, De Waard MA, De Wit PJGM. Contemplations and speculations on novel approaches in the control of fungal plant diseases. Pest. Sci. 1983; 14: 272-293.
- Franich RA, Gadgil PD, Shain L. Fungistaxic effects of Pinus radiata needle epicuticular fatty and resin acids on Dothistroma pini. Physiol. Plant Path. 1982; 23: 183-195.
- He W, Gavai A, Huang FC, Regan J, Hanney B, Poli G, et al. Novel cytokine release inhibitors. Part IV: analogs of podocarpic acid. Bioorg Med Chem Lett. 1999; 9: 469-474.
- Singh SB, Ondeyka JG, Liu W, Chen S, Chen TS, Li X, et al. Discovery and development of dimeric podocarpic acid leads as potent agonists of liver X receptor with HDL cholesterol raising activity in mice and hamsters. Bioorg Med Chem Lett. 2005; 15: 2824-2828.
- Snider BB, Mohan RJ, Kates SA. Manganese (III)-based oxidative free-radical cyclization. Synthesis of (±)-Podocarpic Acid. J. Org. Chem. 1985; 50: 3659-3661.
- Welch SC, Hagan CP, Kim JH, Chu PS. Stereoselective total syntheses of diterpene resin acids. J. Org. Chem. 1977; 42: 2879-2887.
- Gisvold O, Wilson CO, Doerge RF, Daniels TC. Organic Medicinal and Pharmaceutical Chemistry. Philadelphia, PA, USA. 1971.
- Niederl JB, Vogel HJ. Estracatechol. J. Am. Chem. Soc. 1949; 71: 2566-2568.
- Holoway C, Werbin H. Partial degradation of the benzene ring of estrone; isolation of carbon atoms 2 and 4. J Biol Chem. 1956; 223: 651-660.
- Utne T, Jobson RB, Babson RD. Synthesis of 2- and 4-fluoroestradiol. J. Org. Chem. 1968; 33: 2469-2473.
- Santaniello E, Ravasi M, Ferraboschi P. A-Ring nitration of estrone. J. Org. Chem. 1983; 48: 739-740.
- Parish EJ, McKeen GG, Miles DH. Low temperature ozonolysis of methyl o-methyl podocarpate. Org. Prep. Proc. Int. 1985; 17: 143-146.
- King LC, Ostrum GK. Selective Bromination with Copper (II) Bromide. J. Org. Chem. 1964; 29: 3459-3461.
- Kosower EM, Cole WJ, Wu GS, Cardy DE, Meisters G. Halogenation with Copper (II). I. Saturated Ketones and Phenol. J. Org. Chem. 1963; 28: 630-633.
- Lickel AE, Rieke AC, Wheeler DM. Synthesis of diterpenoid acids. VI. Conformations of some derivatives of podocarpic acid. J Org Chem. 1967; 32: 1647-1649.
- Cutfield JF, Waters TN, Clark GR. Conformations of diterpenoids: an X-ray determination of the molecular structure of 6a-bromo-13-hydroxy-14-isopropylpodocarpa-8,11,13-trien-7-one and of the structure and absolute configuration of methyl 6a-bromo-13-isopropyl-7-oxopodocarpa-8,11,13-trien-15-oate. J. Chem. Soc. Perkin Trans 2. 1974; 150-157.
- Cambie RC, Clark GR, Crump DR, Waters TN. The stereochemistry of 6-bromo-7-oxoditerpenoids. Chem. Comm. 1968; 183-185.
- Clark GR, Waters TN. Crystal structure and absolute configuration of the diterpenoid, methyl 6a-bromo-12-methoxy-7-oxopodocarpate. J. Chem. Soc. C. 1970; 887-892.
- Wenkert E, Beak P, Carney RWJ, Chamberlin JW, Johnston DBR, Roth CD, et al. Derivatives of dehydroabietic and podocarpic acids. Can. J. Chem. 1963; 41: 1924-1936.
- Bose AK, Manhas MS, Cambie RC. Rotatory Dispersion of Some Brominated a-Tetralones. J. Org. Chem. 1965; 30: 501-504.
- Wenkert E, Fuchs A, McChesney JD. Chemical Artifacts from the Family Labiatae. J. Org. Chem. 1965; 30: 2931-2934.
- Miles DH, Parish EJ. Inducement of one-step dehydrobromination-decarbomethoxylation and o-alkyl cleavage of methyl esters by 1,5-diazabicyclo[4.3.0]nonene-5(DBN). Tetrahedron Lett. 1972; 13: 3987-3990.
- Parish EJ, Miles DH. O-Alkyl cleavage of methyl esters by 1,5-diazabicyclo[5.4.0]undecene-5. J. Org. Chem. 1973; 38: 1223-1225.
- Parish EJ, Mody NV, Hedin PA, Miles DH. Cleavage of d-Keto β, g-Unsaturated Esters by 1,4-Diazabicyclo[2.2.2]octane. J. Org. Chem. 1974; 39: 1592-1593.
- Parish EJ, Haung BS, Miles DH. Selective Cleavage of β-Keto and Vinylogous β-Keto Esters by 3-Quinuclidinol. Synth. Comm. 1975; 5: 341-345.
- Parish EJ. Synthetic approaches to antifungal terpenoids. Reactions of amine bases [dissertation]. Mississippi State University. 1974.
- Parish EJ, Honda H, Chitrakorn S, Livant P. A facile chemical synthesis of cholest-4-en-3-one. Carbon-13-nuclear magnetic resonance spectral properties of cholest-4-en-3-one and cholest-5-en-3-one. Lipids. 1991; 26: 675-677.
- Karplus M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963; 85: 2870-2871.
- Parish EJ. Analysis of Oxysterols. In: Analysis of Sterols and Other Biologically Significant Steroids. Nes WD, Parish EJ, editors. Academic Press. 1989; 133-149.
- Bhacca NS, Williams DH. Applications of NMR Spectroscopy in Organic Chemistry. San Francisco, CA, USA. 1964.
- Levy GC, Lichter RL, Nelson GL. Carbon-13 Nuclear Magnetic Resonance Spectroscopy. 2nd edn. New York: Wiley-Interscience. 1980.
- Silverstein RM, Bassler GC, Morrill TC. Spectrometric Identification of Organic Compounds. 3rd edn. New York: John Wiley & Sons. 1974.
- Benn R, Gunther H. Modern pulse methods in high-resolution NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 1983; 22: 350-380.
- Hikichi K. Strategy for molecular structure analysis using two-dimensional NMR. JEOL News, Anal. Instr. 1984; 20: 10.
- Burnell RH, Jean M, Savard S. Synthesis of coleon B tetramethyl ether. Can. J. Chem. 1983; 61: 2461-2465.
- Duddeck H, Dietrich W. Structure Elucidation by Modern NMR. New York: Springer-Verlag. 1989.
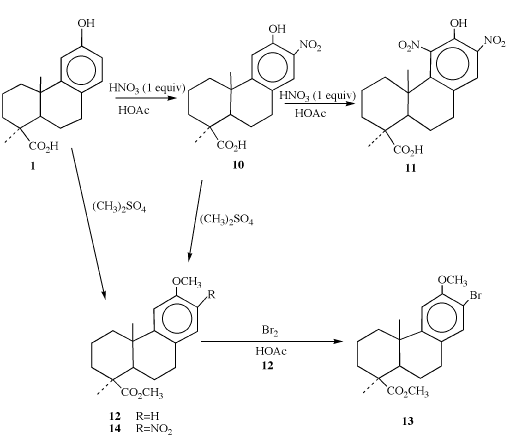
Scheme 1
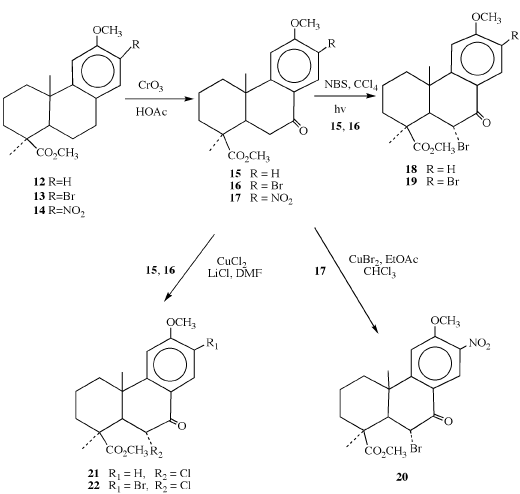
Scheme 2
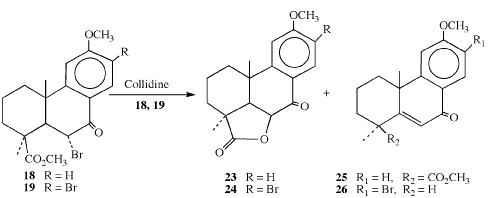
Scheme 3
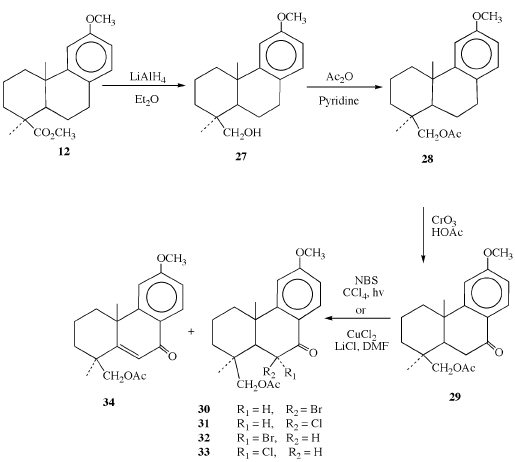
Scheme 4
The first modification, substitution of the electron-withdrawing halogen and nitro groups onto C (11) and/or C (13) of the aromatic ring, was based upon the well-known observation that the antiseptic properties of phenols are enhanced by the introduction of these groups onto the phenolic ring [27].
Nitration was accomplished by reacting podocarpic acid 1 (Scheme 1) with nitric acid in acetic acid [28-31]. The number of nitro groups introduced onto the aromatic ring was controlled by the amount of nitric acid used in the reaction (one or two equivalents, 10 or 11). The 13-nitro derivative 10 was methylated with dimethyl sulfate under basic condition to yield 14. A similar methylation of 1 has been shown to produce methyl O-methyl podocarpate 12 [4,15]. Bromine was introduced at C-13 by the electrophilic substitution of bromine into the aromatic ring of 12 using bromine in acetic acid. The fact that this reaction gives only the monosubstituted 13-bromo derivative is probably due to steric hindrance resulting from the angular methyl group in the axial orientation at position 10 and the large size of the bromine atom which would prevent substitution at position 11, the other ortho position on the ring.
In scheme 2, benzylic oxidation of 12, 13, and 14 using chromium trioxide produced the corresponding ketone derivatives (16, 17). It has also recently been shown that 12 may be oxidized to the ketone 15 under conditions of ozonolysis [32]. Ketones 15 and 16 were brominated using an adapted procedure derived from the work of Bible and Grove [4,15] to yield the mono- and di- bromoketones 18 and 19. In order to effect the bromination of 17, an alternate method was utilized which gave ample quantities of bromoketone 20 [33]. The corresponding chloride derivatives (21 and 22) of 15 and 16 were prepared by reaction with copper chloride and lithium chloride in N, N-dimethylformamide [34]. The assignments of the α- configuration to the halogen atoms at C-6 were verified using known coupling constants from the 1H NMR spectra which are correlated to the X-ray structure determination of 18 [4,35-39,40].
In scheme 3, the α-bromoketones 18 and 19 were converted to lactones 23 and 24 by refluxing in collidine [4,15,39]. By-products of this reaction include the α, β-unsaturated ketone 25 which results from the dehydrobromination [39,41] of 18. Ketone 26 results from a one-step dehydrobromination-decarbomethoxylation [42-45] of 19. The synthesis of 18, lactone 23 and ketone 25 are not reported here and were made for use in the identification of the corresponding products 19, 24 and 26. The synthesis of products 18, 24, and 26 has been described previously and are noted above.
The acetate series of compounds (scheme 4) was synthesized by hydride reduction of methyl O-methyl podocarpate 12 followed by acetylation of the resulting alcohol 27 [46]. The acetate 28 was then oxidized at the benzylic position to ketone 29. In contrast to the methyl ester derivatives, halogenations at position 6 (using methods described previously) of the corresponding keto acetates resulted in two epimers, the 6 α- and 6 β- halogenated compounds, as well as the dehydrohalogenation product 34. The assignments of the α- and β-configuration to the halogen atoms at C-6 were determined from the 1H NMR coupling constants at C-5 and C-6 as described previously.
Experimental
General methods
Procedure for recording of melting points (m.p) and infrared (IR), 1H NMR, and mass (MS) spectra were those used previously [47]. Similarly, details concerning the use of thin-layer (TLC) and column chromatography have been described [47]. Solvent systems for TLC analysis were: 20% ether in toluene (by volumes) in all cases unless stated otherwise. Elemental analyses were performed by Galbraith Laboratories, Knoxville, Tennessee, USA.
The observed stereochemistry of substituents and ring substitution patterns were similar to those recorded in previous studies using related terpenoids and steroids (not in the podocarpic acid series) by observing coupling constants and dihedral angles and applying the Karplus equation [36-38,48-55].
Chemical synthesis of podocarpic acid derivatives
Podocarpic Acid (1) Pure podocarpic acid 1 was used as the starting material for the synthesis of the nitro and dinitro derivatives and was obtained by recrystallization of 500 g of the crude natural product from methanol and water after filtration from hot methanol to remove extraneous material such as twigs and bark. Pure 1 was obtained (394 g, 79%) as beige crystals (Rf=0.25) having mp 186-l88°C. (lit. mp 187-188°C55). The yield varied, of course, with the amount of extraneous material present in the crude natural product (recrystallization of 1000 g crude material yielded 870 g of pure 1). IR (KBr): 3327, 1695, 1500, and 1180 cm-l; lH NMR (acetone-d6); δ1.10 {3H, s, C(15)-H3}, 1.29 {3H, s, C(17)-H3}, 6.56 (lH, dd, J=2.4, 8 Hz, aromatic), 6.76 (lH, d, aromatic, J=8.0 Hz), 6.83 (lH, d, aromatic, J=2.4 Hz), and 8.0 (lH, broad singlet, exchangeable, phenolic -OH); LRMS: m/e 274 (M, 100%), 259 (M-CH3, 36%), 213 (85%), and 157 (24%).
13-Nitro Podocarpic Acid (10) Pure podocarpic acid 1 (10 g, 0.036 mol) was dissolved in 250mL glacial acetic acid (45°C). This solution was cooled to 35°C and 2.5mL (1 equiv) concentrated nitric acid was added dropwise with stirring, turning the brownish solution a deep red color. After about five minutes, a bright yellow precipitate began to form. Stirring was continued for three hours, the solution was filtered, the precipitate washed with water and recrystallized from acetone and water to yield 8.5 g (74%) of the mononitro derivative 10 as a bright yellow powder having Rf=0.45 and mp 160°C(d). IR (KBr): 3259 (broad), 1686, 1626, 1569, 1518, 1473, 1420, 1318, and 1257 cm- 1; lH NMR (acetone-d6): 1.18 {3H, s, C(15)-CH3}, 1.32 {3H, s, C(17)- CH3}, 7.11 (lH, s, aromatic), and 7.79 (lH, s, aromatic); MS: m/e 319 (M, 98.8%), 304 (M-CH3, 12.1%), 258 (100%), and 202 (15.4%). Anal. Calc'd: C, 63.94%; H, 6.63%. Found: C, 64.00%; H, 6.60%.
11,13-Dinitro Podocarpic Acid (11) The dinitro derivative 11 was prepared by the same procedure as the mononitro derivative 10, with the addition of 7.5mL (3 equiv) of concentrated nitric acid to 10 g of 1 rather than 1 equiv. The resulting solution turns a lighter shade of red and no precipitate is formed until the reaction mixture is poured into water. The 11,13-dinitro derivative 11 (6.8 g, 52%) isolated is a powder and is a lighter shade of yellow than the mononitro derivative. This product was recrystallized from methanol and water to yield 5.4 g (47%) of 11 having Rf=0.30 and mp 2l3-215°C. IR (KBr): 3193 (broad), 1694, 1622, 1578, 1546, 1458, 1423, 1313, and 1262 cm-l; lH NMR (acetone-d6): δl.33 {3H, s, C(15)-H3}, 1.34 {3H, s, C(17)-H3}, and 8.02 {lH, s, C(14)-H}; LRMS: m/e 364 (M, 23.9%), 346 (M-H20, 29.9%), 328 (42.4%), 304 (M-2NO, 12.0%), 303 (42.6%), 129 (44.5%), 128 (62.6%), 116 (41.8%), 115 (79.4%), 91 (64.5%), 77 (78.5%), and 55 (100%). Anal. Calc'd: C, 56.04%; H, 5.53%. Found: C, 56.10%; H, 5.50%.
Methyl O-Methyl l3-Nitropodocarpate (14) 10 g (0.31 mol) 13-nitro podocarpic acid 10 was dissolved in 250mL methanol and 150mL 10% potassium hydroxide. To this solution was added 130mL dimethyl sulfate in 5mL aliquots over a period of four hours with continuous stirring. The solution was kept in a cold water bath and the temperature not allowed rising above 38°C. After 60mL of the dimethyl sulfate had been added, a yellow precipitate had begun to form and the solution was stirred 45 minutes before continuing the addition. To obtain a pure product in good yield, it was imperative that the reaction mixture be kept alkaline by the addition of aliquots of 40% potassium hydroxide as needed during addition of the dimethyl sulfate. The pH is easily monitored, as the solution is deep red when alkaline and gradually goes from orange to bright yellow as the pH becomes acidic. After dimethyl sulfate addition, the reaction mixture was kept at 0°C overnight. The solution was filtered and the yellow precipitate washed first with 5% KOH and then with water to yield 9.7 g (90%) of 14 (Rf=0.76, mp 90-92°C). IR (KBr): 1708, 1664, 1569, 1511, 1451, 1335, 1272, 1223, 1195, 1148, and 1033 cm-l; 1H NMR (CDCl3): 1.05 {3H, s, C(15)-H3}, 1.28 {3H, s, C(17)-H3}, 3.65 (3H, s, ester -OCH3), 3.87 (3H, s, ether -OCH3), 6.92 (lH, s, aromatic), and 7.50 (lH, s, aromatic); MS: m/e 347.7 (M, 94.4%), 332.6 (M-CH3, 3.4%), 272.7 (M-C6H3 100%), and 206.6 (11.5%). Anal. Calc'd: C, 65.69%; H, 7.25%. Found: C, 65.85%; H, 7.18%.
Methyl O-Methyl l3-Bromopodocarpate (13) Methyl ester 12 (1.0 g, 0.0033 mol) was dissolved in 20mL glacial acetic acid and 1.1 g bromine (0.36mL, 0.0066 mol) in 20mL glacial acetic acid was added dropwise. The solution was allowed to stand ten minutes at room temperature before it was poured over ice. The resultant crystals were filtered, washed with water and recrystallized from acetone and water to yield 1.14 g (90%) of iridescent crystals of 13 having mp 138- 140°C. IR (KBr): 1720, 1590, 1485, and 1450 cm-l ; 1H NMR (CDCl3): δl.15 {3H, s C(15)-H3}, 1.33 {3H, s C(17)-H3}, 3.65 (3H, s, ester -OCH3), 3.82 (3H, s, ether -OCH3), 6.77 {lH, s, C(ll)-H}, and 7.19 {lH, s, C(14)-H}; MS: m/e 382 and 380 (M, 96.6%, 100%), 367 and 365 (M-CH3, 6%, 3%), 307 and 305 (M-C6H3, 63%, 62%), and 226 (18%). Anal. Calc'd: C, 59.85%; H, 6.61%. Found: C, 59.65%; H, 6.56%.
Methyl O-Methyl Podocarpate (l2) Crude podocarpic acid 1 (250 g) was dissolved in 500mL methanol, 250 g ice, and 120 g sodium hydroxide and stirred 90 minutes. The dark brown solution was cooled to 15OC and 215mL dimethyl sulfate added in 10mL portions, with stirring, over a 2-hour period. The reaction mixture was kept in an ice bath to avoid overheating of the exothermic reaction. The mixture began to solidify after the addition of 80mL dimethyl sulfate and after the addition was completed, was allowed to warm to room temperature. After standing overnight, 2 liters (L) of water were added to the solidified mass which was then filtered on a Buchner funnel and washed with water. The light-brown residue was then warmed in water on a steambath and refiltered and recrystallized from acetone and water (filtration of the hot acetone solution before the addition of water removed bark and twigs) to yield 201 g of 12 as a fluffy white water-insoluble solid, Rf=0.89, mp 124-126°C, (lit mp1 128°C). IR (KBr): 1710, 1605, 1500, 1465 cm-1; 1H NMR (CDCl3): 1.05 {3H, s, C(15)-H3}, 1.29 {3H, s, C(17)-H3}, 3.66 {3H, s, ester -OCH3}, 3.78 {3H, s, ether -OCH3}, 6.82 {3H, m, ArH}; MS: m/e 302 (M, 96%), 227 (M-C6H3, 100%), 171 (9%), and 161 (5%).
Methyl O-Methyl 7-Ketopodocarpate (15) 100 g (0.33 mol) methyl O-methyl podocarpate 12 was dissolved in 1 L glacial acetic acid. 94 g chromium trioxide (0.93 mol) was dissolved in 1 L 8O% acetic acid and this mixture added slowly, with stirring (reaction is exothermic), to the methyl o-methyl podocarpate solution. This mixture was allowed to stand overnight and poured into 6 L saturated sodium chloride solution and again left to stand overnight. The precipitate was removed by filtration and washed well with water. The yellow crystalline solid had mp 124-126°C (lit. mp 121-123°C40, 122-124°C4) and Rf=0.76 (50% ether in toluene). If a greenish color persisted from the reaction, the ketone was recrystallized from acetone and water to give 75 g (71.6%) of 15. IR (KBr): 1720, 1670, and 1595 cm-1; lH NMR (CDCl3): δ1.11 {3H, s, C(15)-H3}, 1.26 {3H, s, C(17)-H3}, 3.10 {2H, m, C(6)-H2}, 3.70 {3H, s, ester -OCH3}, 3.86 {3H, s, ether -OCH3}, 6.82 {lH, d, C(13)-H, J=8.7 Hz}, 6.87 {lH, s, C(11)-H}, and 8.02 {lH, d, C(14)-H, J=8.0 Hz}; MS: m/e 316 (M, 57%), 241 (M-C6H3, 26%), 201 {10%), and 190 (12%).
Methyl O-Methyl 13-Bromo-7-ketopodocarpate (l6) Brominated methyl ester 13 (1.0 g, 0.0026 mol) was dissolved in 20mL glacial acetic acid. To this solution was added a solution of 0.8 g (0.0079mol) chromium trioxide in 80% acetic acid. The resultant mixture was allowed to stand in a stoppered flask at room temperature overnight before being poured into 60mL saturated sodium chloride. After twenty-four hours, the resulting precipitate was filtered, washed with water, and recrystallized from acetone to yield 0.7 g (69%) of fluffy white crystals of 16, mp 175-178°C. IR (KBr): 1720, 1665, 1586, 1486, and 1438 cm-l; lH NMR (CDCl3): δ1.12 {3H, s, C(15)-H3}, 1.27 {3H, s, C(17)-H3}, 3.10 {2H, q, C(6)-H2}, 3.71 (3H, s, ester -OCH3), 3.96 (3H, s, ether OCH3), 6.84 {lH, s, C(ll)-H}, and 8.22 {lH, s, C(14)-H}; MS: m/e 396 and 394 (M, 100%, 87%), 382 and 380 (M-CH3, 78%, 75%), 365 (M-OCH3, 3.75%), 321 and 319 (M-C6H3, 56%, 53%), 307 and 305 (52%, 46%), 253 (23%), 226 (13%), 213 (16%), and 101 (18%). Anal. Calc'd: C, 57.73%; H, 5.86%. Found: C, 57.58%; H, 5.79%.
Methyl O-Methyl 6α,13-Dibromo-7-ketopodocarpate (19) Bromoketone 16 (7.0 g, 0.018 mol) was dissolved in 157mL carbon tetrachloride and 4.0 g N-bromosuccinimide was added. This mixture was placed in direct sunlight for four days. The mixture was filtered, the residue washed with cold carbon tetrachloride and the filtrate evaporated in vacuo. The resulting residue was recrystallized from methanol and water three times to yield 6.0 g (75%) of 19 as a white crystalline solid having Rf=0.70 and mp 181.5-183°C. IR (KBr): 1717, 1671, and l581 cm-1; 1H NMR (CDCl3): δ0.86 {3H, s, C(15)-H3}, 1.56 {3H, s, C(17)-H3}, 2.51 {1H, d, C(5)-H, J=7.1 Hz}, 3.74 (3H, s, ester -OCH3), 3.97 (3H, s, ether -OCH3), 5.82 {1H, d, C(6)-H, J=7.0 Hz}, 6.82 {1H, s, C(ll)-H}, and 8.01 {lH, s, C(14)-H}; MS: m/e 476, 474, 472 (M, 10.2%, 17.7%, 9.7%), 396, 395, 394, and 393 (M-H81Br and M-H79Br, 38.1%, 100%, 55.8%, and 97.1%), 335 and 333 (58.6% and 55.7%), 321 and 319 (36.2% and 45.1%), 101 (41.0%), and 82 and 80 (H81Br and H79Br, 25.9% and 26.0%). Anal. Calc'd: C, 48.13%; H, 4.68%. Found: C, 48.25%; H, 4.71%.
13-Bromo Podolactone (24) and Unsaturated Ketoester (26). The 13-Bromo Podolactone 24 from 4 g (0.0093 mol) of 19 was formed in refluxing collidine by the same procedure used to form lactone 23 from 25. In addition to the 13-bromo podolactone 24 (0.4 g, 10%) the 13-bromo unsaturated ketoester 26 (0.4 g, 10%) was isolated. These products were separated by column chromatography on silica gel using solvent gradient of 0-25% ether in toluene and characterized. The Podolactone 24 was a white solid having mp 153- 155°C. IR (KBr): 1775, 1683, 1582, 1287, 1037, and 674 cm-l; lH NMR (CDCl3): δ1.13 {3H, s, C(15)-H3}, 1.36 {3H, s, C(17)-H3}, 2.34 {1H, d, C(5)-H, J=5.7 Hz}, 3.99 (3H, s, C(12) -OCH3}, 4.92 (1H, d, C (6) -H, J=5.58 Hz}, 6.74 {lH, s, C(ll)-H}, and 8.04 {lH, s, C(14)-H}; LRMS: m/e 380 and 378 (M+, 43.3 and 41.7%), 365 and 363 (M+-CH3, 5.5 and 5.4%), 352 and 350 (M+-CO, 4.0 and 4.0%), 336 and 334 (M+-CO2, 3.5 and 4.3%), 323 and 321 (16.0 and 20.6%), 309 and 307 (13.4 and 16.0%), 255 and 253 (97.1 and 100%), 174 (14.9%), and 110 (22.2%), 70 (15.4%), 57 (15.4%), and 55 (16.1%). Anal. Calc'd: C, 57.01%; H, 4.97%. Found: C, 56.89%; H, 5.10%. The 13-bromo unsaturated ketoester 26 was characterized by NMR. lH NMR (CDCl3): δ1.33 {3H, s, C(15)-H3}, 1.49 {3H, s, C(17)-H3}, 3.65 {3H, s, ester -OCH3), 3.97 (3H, s, ether -OCH3), 6.57 ({1H, s, vinylic C(6) -H}, 6.92 {lH, s, C(ll)-H}, and 8.35 {lH, s, C(14)-H}, (characterized by comparison with a sample of ketone 25).
Methyl O-Methyl 6α-Chloro-7-ketopodocarpate (21) Copper (II) chloride dihydrate (9.0 g) and lithium chloride (1.6 g) were dissolved in 30mL N,N-dimethylformamide before the addition of 5.0 g (0.016 mol) ketone 15. The reaction mixture was heated at 90-100OC for six hours and then allowed to cool before the addition of 60mL water, followed by ether extraction. The organic layer was dried over anhydrous magnesium sulfate and evaporated in vacuo. Recrystallization of the residue from methanol and water gave 4.5 g (80%) of 21 as a white crystalline solid (Rf=0.64) with mp 108-l09OC. IR (KBr): 1710, 1680, and 1600 cm-l; 1H NMR (CDCl3): δ0.87 {3H, s, C(15)-H3}, 1.48 {3H, s, C(17)-H3}, 2.28 {1H, d, C(5)-H, J=8 Hz}, 3.71 {3H, s, ester -OCH3}, 3.87 {3H, s, ether -OCH3}, 5.69 {lH, d, C(6)-H, J=7.9 Hz}, 6.50 {2H, m, Ar-H}, and 7.83 {lH, d, C(14)-H, J=8.4 Hz}; MS: m/e 352 and 350 (M, 25.3%, 72.9%), 315 (M-H37Cl and M-H35Cl, 23.9%), 255 (48.6%), 227 (19.8%), 201 (30.2%), and 175 (100%). Anal. Calc'd: C, 65.05%; H, 6.61%. Found: C, 64.99%; H, 6.50%.
Methyl O-Methyl 6α-Chloro-13-Bromo-7-ketopodocarpate (22) Bromoketone 9 (5.0 g, 0.013 mol) was chlorinated using the same procedure used to chlorinate 15 to form 21. 4.2 g (75%) of 22 was formed as a white crystalline solid having mp 158-159.5°C. IR (KBr): 1712, 1689, 1583, and 1458 cm-l; lH NMR (CDCl3): δ0.89 {3H, s, C(15)-H3}, 1.48 {3H, s, C(17)-H3}, 2.28 {lH, d, C(5)-H, J=8.0 Hz}, 3.72 (3H, s, ester -OCH3), 3.96 (3H, s, ether -OCH3), 5.68 {lH, d, C(6)-H, J=7.8 Hz}, 6.85 {lH, s, C(ll)-H}, and 8.01 {lH, s, C(14)-H}; MS: m/e 432, 430, and 428 (M, 27%, 100%, and 72%), 335, 334, and 333 (28%, 18%, and 35%), and 255 and 253 (44% and 48%). Anal. Calc'd: C, 53.10%; H, 5.16%. Found: C, 53.00%; H, 4.92%.
Methyl O-Methyl 13-Nitro-7-ketopodocarpate (17) The same procedure was used to prepare 17 from 10 g (0.029 mol) of 14 as was used in the oxidation of 12 to 15 with the exception that the reaction mixture was placed on a steambath for twenty-four hours and the precipitate was filtered and washed with acetic acid and water before the supernatant was poured into saturated sodium chloride. The initial precipitate was pure 16 and a slightly impure second crop was obtained from the supernatant which was recrystallized from acetone and water to give a total of 7.5 g (72%) of 17 as a bright yellow crystalline solid having Rf=0.50 and mp 255-258°C. IR (KBr): 1720, 1677, 1606, 1517, 1347, 1313, 1287, 1260, 1231, 1187, 1165, 1146, and 1028 cm-1; 1H NMR (CDCl3): δ1.15 {3H, s, C(15)-H3}, 1.28 {3H, s, C(17)-H3}, 3.15 {2H, m, C(6)-H2}, 3.70 (3H, s, ester -OCH3), 3.99 (3H, s, ether -OCH3), 7.00 {lH, s, C(ll)-H}, and 8.45 {lH, s, C(14)-H}; LRMS: m/e 361 (M, 100%), 329 (M-CH2OH, 10%), 301 (M-CO2CH3, 33%), 286 (M-C6H3, 40%), 260 (14%), 246 (30%), 235 (48%), 220 (65%), 101 (32%), 95 (39%), and 69 (45%). Anal. Calc'd: C, 63.15%; H, 6.42%. Found: C, 63.22%; H, 6.45%.
Bromination of O-Methyl 7-Ketoacetate (29). The procedure was the same as that used for the α-bromination of 15 to form 18. TLC analysis showed three major bands having R, values of 0.71, 0.67, and 0.47. After separation by column chromatography on silica gel in 0-25% ether in toluene and recrystallization from acetone and water, the band having Rf=0.71 was shown to be 6β-bromo-7-ketoacetate 30 in the form of yellowish crystals (30%), mp 135.5-137°C; the band having Rf=0.67 was the desired 6α-bromo-7-ketoacetate 32 in the form of colorless crystals (25%), mp 105-106°C and the band having Rf=0.47 was the elimination product 34 (25%). The spectral data for each of the three products are given below.
O-Methyl 6α-Bromo-7-Ketoacetate (32). IR (KBr): 1726, 1672, 1600, 1284, 1246, 1037, and 1019 cm-1; 1H NMR (CDC13): 1.07 {3H, s, C(l5)-H3}, 1.36 {3H, s, C(l7)-H3}, 2.15 (3H, s, CO-CH3), 2.41 {lH, d, C(5)-H, J=8.2 Hz}, 3.88 (3H, s, ether -OCH3), 4.32 (2H, s, CH2- OAc), 5.01 {lH, d, C(6)-H, J=8.1 Hz}, 6.80 {lH, d, C(l1)-H, J=2.4 Hz}, 6.87 {lH, q, C(13)-H}, and 7.79 {1H, d, C(14)-H, J=8.5 Hz}; LRMS: m/e 410 and 408 (M, 16.8%, 16.9%), 330 (M-Br, 22.1%), 287 (21.3%), 269 (33.6%), 258 (20.3%), 255 (28.6%), 241 (41.4%), 227 (34.2%), 201 (91.0%), 175 (100%), 82 and 80 (H79Br and H81Br, 57.1% and 60.4%). Anal. Calc'd: C, 58.69%; H, 6.16%. Found: C, 58.60%; H, 6.00%.
O-Methyl 6β-Bromo-7-ketoacetate (30). IR (KBr): 1735, 1663, 1592, 1270, 1243, and 1022 cm-1; 1H NMR (CDC13): 1.18 {3H, s, C(l5)-H3}, 1.77 {3H, s, C(l7)-H3}, 2.10 (3H, s, CO-CH3), 2.29 {lH, d, C(5)-H, J=2.7 Hz}, 3.88 (3H, s, ether -OCH3), 4.76 (2H, q, -CH2OAc), 4.93 {lH, d, C(6)-H, J=2.7 Hz}, 6.87 (2H, m, Ar-H), and 8.12 {lH, d, C(l4)-H, J=9.4 Hz}; LRMS: identical to 31 except m/e 82 and 80 (10.9% and 11.5%). Anal. Calc'd: C, 58.69%; H, 6.16%. Found: C, 58.65%; H, 6.26%.
Unsaturated 7-Ketoacetate 34. 1H NMR (CDCl3): δ 1.30 {3H, s, C(l5)-H}, 1.46 {3H, s, C(l7)-H}, 2.05 (3H, s, CO2-CH3), 3.88 (3H, s, ether OCH3), 4.20 (2H, q, CH2-OAc), 6.48 {lH, s, vinylic C(6)-H}, 6.92 {lH, d, C(ll)-H, J=8.7 Hz}, 6.95 {lH, d, C(l3)-H}, and 8.11 {lH, d, C(l4)-H}, (characterized by comparison with a sample of ketone 25).
Chlorination of O-Methyl 7-Ketoacetate (29) The procedure was the same as that used to α-chlorinate 15 to form 21. TLC analysis indicated the desired O-methyl 6α-chloro-7-ketoacetate 33 to be the major product when 1 g (0.0030 mol) 7-ketoacetate 29 was reacted, contaminated by small amounts of 6β-chloro-7-ketoacetate 31 and the elimination product, unsaturated 7-ketoacetate 34. These minor products could be removed by column chromatography on silica gel in 0-3% ether in toluene or by recrystallization from methanol and water to yield 0.78 g (70%) of O-methyl 6α-chloro-7-ketoacetate 33 as pale yellow crystals having Rf=67 and mp 119-120°C. However, when the reaction was scaled up to 5 or 10 g, several products were seen on TLC and 33 was no longer the major product. Attempted recrystallization from methanol and water yielded only oil which was also seen to be a mixture of several products when analyzed on TLC. For this reason, the reaction was run in several 1 g batches. The spectral data for the 6α- and 6-chloro compounds 33 and 31 are reported below.
O-Methyl 6α-Chloro-7-ketoacetate (33) IR {KBr): 1726, 1676, 1596, 1291, 1249, 1210, 1037, and 1021 cm-1; 1H NMR {CDC13}: δ 1.15 {3H, s, C(l5)H3}, 1.30 {3H, s, C(l7)-H3}, 2.15 (3H, s, CO-CH3), 2.25 {lH, d, C(5)-H, J=9.5 Hz}, 3.88 (3H, s, ether -OCH3), 4.36 (2H, q, CH2-OAc), 4.89 (lH, d, C(6)-H, J=9.5 Hz}, 6.85 (2H, m, Ar-H), and 7.87 {lH, d, C(l4)-H, J=9.5 Hz}; LRMS: m/e 366 and 364 (M, 29.7% and 10.3%), 330 (7.4% ), 328 (M-H35Cl), and (M-H37Cl, 4.4%), 269 (M-CH3CO2Cl, 19.3%), 201 (32.2%), and 175 (100%). Anal. Calc'd: C, 65.84%; H, 6.91%. Found: C, 65.79%; H, 7.05%.
O-Methyl 6β-Chloro-7-ketoacetate (31).1H NMR (CDCl3): δ 1.18 {3H, s, C(l5)-H3}, 1.73 {3H, s, C(l7)-H3}, 2.06 (3H, s, CO-CH3), 3.88 (3H, s, ether -OCH3), 4.83 {lH, d, C(6)-H, J=3.0 Hz}, 6.85 (2H, m, Ar-H), and 8.00 {lH, d, C(l4)-H, J=9.5 Hz}.
Conclusion
In conclusion, these studies have demonstrated that podocarpic acid is capable of extensive chemical modification into a library of diverse compounds. The basic podocarpic acid structure has yielded a variety of derivatives, which can be evaluated for potential biological activity for use in medicine and agriculture.
Acknowledgement
Support for WCN provided by the Defense Threat Reduction Agency under Basic Research Grant HDTRA1-10-1-0004 is gratefully acknowledged.
References