
Research Article
Austin J Biosens & Bioelectron. 2016; 2(1): 1019.
DNA Methylation and miRNA Profiling of NSC Differentiation on Electrospun POMA Nanofibers
Augustus T Mercado1,2, Yui Whei Chen-Yang2,3, Ting Yu Chin1,3, Jui-Ming Yeh2,3* and Chung-Yung Chen1,3
¹Department of Bioscience Technology, Chung Yuan Christian University, Taiwan
²Department of Chemistry, Chung Yuan Christian University, Taiwan
³Center for Biomedical Technology, Chung Yuan Christian University, Taiwan
*Corresponding author: Jui-Ming Yeh, Department of Chemistry and Chung Yung Chen, Department of Bioscience Technology, Chung Yuan Christian University, No. 200, Chung-Pei Road, Chung-Li, 32023, Taiwan
Received: June 09, 2016; Accepted: July 20, 2016; Published: July 25, 2016
Abstract
A genetic level assessment of Neural Stem Cell (NSC) differentiation towards a synthesized novel nanofibrous material is needed for its biomedical applications. NSCs differentiated on a neat electrospun Poly-O-Methoxyaniline (POMA) underwent a modified method of PCR Selective Suppression Hybridization (PSSH) to provide a DNA methylation profile. DNA fragments were sequenced and identified using BLAST. Then, identified genes were annotated using Gene Set Toolkit (Gestalt), Database for Annotation, Visualization and Integrated Discovery (DAVID) and Pathway Interaction Database (PID). Eleven genes (NCAM2, KIF5C, DMD, ELMO1, F2R, FZR1, HCN1, DAGLA, HSPB8, RAB38, and CMKLR1) corresponding to neural stem cell functions were methylated after differentiation on the nanomaterial. This indicates that POMA scaffold was efficient in enhancing differentiation of NSCs. Moreover, the miRNA profiles of NSC before and after differentiation were assessed by miRNA microarray. POMA nanofibers stimulated the expression of miR-1224, miR-204- 3p, miR-30c-1-3p, and miR-92-5p, as compared to a flat substrate (PDL).The differential upregulation of these miRNAs further enhanced the differentiation and proliferation potential of POMA as a scaffold for neuronal tissue engineering.
Keywords: NSC differentiation; Poly-o-methoxyaniline; DNA methylation; PCR Selective Suppression Hybridization; miRNA microarray
Introduction
Neural tissue engineering aims to repair neural tissue through the use of biological tools such as normal or genetically engineered cells and creating Extracellular Matrix (ECM) equivalents along with potent synthetic tools such as biomaterial for scaffold design. Recent studies showed that Neural Stem Cells (NSCs) possess a great potential as main seed cells in nerve regeneration because of the potential therapeutic effect to a number of CNS disorders [1]. However, increasing evidence has shown that stem cell development requires a niche, which is a local microenvironment that houses stem cells and regulates their self-renewal and differentiation [2]. For this reason, the physical environment of the cells can affect its differentiation. Advanced nano-based polymers are being considered to be used as a substrate biomaterial for nerve engineering due to the simple fact that the nanostructure mimics more closely the ECM dimensions; thus, useful for promoting cell attachment, migration, and proliferation. Thus, there are a number of studies using different types of material as potential biomaterial for the growth and differentiation of NSCs.
Conducting polymers can be applied as biosensors, scaffolds for tissue engineering, neural probes, drug-delivery devices, and bioactuators [3]. Polyaniline is one of the oldest known conducting polymers that has been often studied due to its beneficial properties as a polymeric nanostructure [4]. Many have studied this material because of its low cost, high electrical conductivity and good environmental stability. A prepared neat electrospun poly(omethoxyaniline) (designated as POMA) was previously characterized [5] and used for efficient neural stem cell differentiation. Results of the cell viability assay, immunofluorescence staining, qRT-PCR and calcium image studies confirmed that POMA showed enhanced NSC attachment and accelerated differentiation [6].
However, stem cell differentiation is controlled by a complex pattern of gene regulation, which is governed by an array of cellular signaling pathways. It is believed that extracellular factors and intracellular process, including epigenetic modification, control cell fate specification and differentiation of NSCs [7]. These modifications include DNA methylation, histone modification and non-coding RNA expression, like miRNAs.
Analysis of methylation states in genomic DNA has provided insights into biological phenomena as disparate as genomic imprinting, human disease, and atypical floral morphologies [8]. Bisulfite treatment can be used to determine the methylation states of individual cytosines in DNA. When bisulfite-treated DNA is amplified by PCR, 5mC on the template strand pairs with guanine on the newly synthesized strand; converted cytosine, which is uracil, pairs with adenine. The methylation patterns of individual DNA molecules therefore can be inferred from the sequences of sub-cloned PCR products. Conversely, differentially methylated DNA fragments can be selected through PCR Selective Suppression Hybridization (PSSH). It is an adapted and modified technique known as Suppression Subtractive Hybridization [9]. PSSH is a PCRbased technique to analyze methylation states in the genomic DNA. This is used to selectively amplify target DNA fragments, which are differentially methylated, and simultaneously suppress non-target DNA amplification. The method is based on suppression PCR effect wherein long inverted terminal repeats attaches to DNA fragments that can selectively suppress amplification of undesirable sequences in PCR procedures.
The DNA methylation and miRNA expression profiles of neural stem cell differentiation towards a synthetic and inorganic microenvironment have never been elaborately discussed. The objective of this study is to identify the DNA methylation changes and miRNA expression levels triggered by extrinsic mechanical signals of POMA for regulating the proliferation and differentiation of NSCs. Genomic DNA of NSCs underwent PSSH to have a DNA methylation profile and identify differentially methylated genes after differentiation. Additionally, the effect of the substrate on the miRNA expression was compared with PDL. The data obtained from this study will help in discovering the related mechanisms associated with NSC differentiation on a biomaterial. These would aid in the assessment of the biocompatibility of the material for neural tissue engineering, thereby its potential for regenerative medicine.
Materials and Methods
Preparation of poly (o-methoxyaniline)
First, 0.164 mol of o-methoxyaniline was added into 400 mL of 2.0 M CaCl2 in HCl and cooled to about 0 °C by stirring in an ice bath. Then, 0.038 mol of ammonium per sulfate, which served as an oxidant, was dissolved in 100 mL of 2.0 M CaCl2 in HCl solution at 0°C and it was then mixed with the o-methoxyaniline/CaCl2/HCl solution by stirring for 12 h. Subsequently, an intense blue-green precipitate of POMA was collected and washed on a funnel fitted with filter paper (90 mm diameter, Advantec No. 7 filter paper). The final product was washed usually five times with 1.0 MHCl (aq) to remove any grey salts. Finally, the precipitate was dedoped by stirring with 500 mL of 1.2 M NH4OH (aq) for 48 h. The Emeraldine Base (EB) of POMA was obtained by filtering and drying at a temperature of 60 °C under vacuum for at least 24 h. The yield of POMA was typically 20%. The EB of the POMA powder was dissolved in a co-solvent system of THF/DMF (50:50 v/v) as an electrospinning solution in the concentration range 1–6 wt%. This solution was placed in a plastic syringe (Terumo, 5 ml), and the POMA fibers were produced by electrospinning, controlling specific parameters (Q, H and V). The electrical field used for electrospinning was generated by a variable high-voltage power supply (Matsusada, AU-40R0.75), which can apply voltages as high as 40 kV. The solution was fed by a syringe pump (KD Scientific Model 200) with the feeding rate tuned for conjugated polymer solutions (Q = 0.02, 0.03 and 0.04 mL min-1). A metallic needle was connected to a high-voltage source set at 15 or 20 kV, and nozzle-to-collector distances of 8, 10, 12 and 14 cm were set to collect the electrospun fiber mat samples. Finally, the collected mat was dried in an oven for 1 h at a temperature of 100 °C.
Cell culture and genomic DNA extraction
Pregnant Sprague-Dawley rats were purchased from the National Laboratory Animal Center (Taiwan, ROC). All animal operations received were in humane care according to the Guidelines for Care and Use of Experimental Animals [10]. This study was also approved by the Animal Research Ethics Board of Chung Yuan Christian University (Taiwan, ROC).
NSCs were isolated from the brains of Sprague-Dawley rat embryos on day 14-15. The embryo brains were dissected, cut in to pieces and treated with digesting solution containing 30 mg/ ml papain, 50 mM EDTA, 2 mg/ml cysteins, and 150 mM CaCl2.
Then, the dissected tissue were treated with DNase I, added into 10% horse serum and collected by centrifugation. The neural stem cells were expanded in Dulbeco’s Modified Eagle Medium (DMEM) supplemented with N2, 20 ng/ml Epidermal Growth Factor (EGF), 10 ng/ml basic Fibroblast Growth Factor (bFGF) and the following antibiotics: 0.5% penicillin and 1% streptomycin. Cells were allowed to grow in a CO2 incubator at 37 °C, 5% CO2 and 95% humidity. The number of live cells was counted by trypan blue exclusion assay in a hemocytometer. The electrospun nanofibers on the block of glass were exposed to UV radiation overnight. The cells were seeded on the nanofibers placed in a petri dish at a density of 1 x 106 cells per dish and cultured with DMEM-F12 medium containing 1% N2 supplement. Cells were allowed to grow and proliferate for two days before 0.5 mM of dcAMP was added. Cell adhesion, proliferation, and differentiation were assessed using a Phase Contrast Light microscope. Cells were harvested after 7 days of differentiation and genomic DNA extraction was done using the Phenol-chloroform method. The genomic DNA was extracted on the seventh day from the cultivated cells with and without dbcAMP. The range of the DNA concentration obtained was between 300-450 ng/μl.
Bisulfite conversion of DNA
Bisulfite treatment was performed using the Qiagen® Epitech Bisulfite kit in accordance to the manufacturer’s protocol. A reaction mixture consisted of 1 ng ~ 2 μg of DNA solution, 85 μL of bisulfite mix, 35 μL DNA protect buffer, and diluted to 140 μL with RNase-free water. Bisulfite DNA conversion was conducted using a thermal cycler (ABITM, GenAmp PCR system 9700) with the following conditions: 5 minutes at 95°C, 25 minutes at 60°C, 5 minutes at 95°C, 85 minutes at 60°C, 5 minutes at 95°C, and 175 minutes at 60°C. Converted DNA was cleaned up using EpiTect® Spin Columns provided with the kit. Uracil residues of bisulfite-converted DNA samples (1~2 μg) were cleaved using 1~5U of USERTM Enzyme NEB® (NEB, M5505S) suspended in 1X T4 ligase buffer and incubated at 37°C in a water bath. Uracils were replaced with biotin-dCTP (InvitrogenTM) by incubation at 37°C for 30 minutes with other dNTPs. The reaction was stopped by incubating the mixture at 75°C for 20 minutes. To reestablish the phosphodiester bonds of double-stranded biotinylated DNA, the mixture was suspended in 1x T4 DNA ligase buffer for a total volume of 70 μl with 350 units of T4 DNA ligase (TAKARA) and incubated at 4°C overnight or longer for successful ligation.
Separation of biotinylated DNAs
Dynabeads®MyOneTM Streptavidin (InvitrogenTM, 650.01) with a stock concentration of 10mg/mL and a binding capacity of ~20μg double-stranded biotin-labeled DNA per mg Dynabeads® was initially washed with 1X BW buffer according to the manufacturer’s instructions. Washings were done repeatedly as deemed necessary using an Easy 50 EasySep® Magnet (STEMCELL TECHNOLOGIES) to separate the supermagentic Dynabeads® from the buffer and preservatives. Dynabeads® were then resuspended in 2X BW buffer to a final concentration of 5μg/μL. For optimum immobilization, an equal volume of biotin-labeled DNA suspended in water is added to the resuspended Dynabeads® to reduce the NaCl concentration in the buffer from 2M to 1M. Incubation was done at room temperature with gentle rotation for 15 minutes. Biotin-labeled DNA bound to Dynabeads® was separated by three-minute incubations in the Easy 50 EasySep® Magnet and washed with 1X BW buffer. Washed supernatant was collected in a separate tube and labeled as methylated DNA from differentiated cells. The immobilized biotin-labeled DNA was dissociated from Dynabeads® by incubation at 90°C in 95% form amide and 10mM EDTA for two minutes. Released biotin-labeled DNA contained the unmethylated DNA from undifferentiated NSCs. Methylated and unmethylated samples were desalted using Amicon® Ultra-0.5 50K centrifugal filters (MILLIPORE Corp.) according to manufacturer’s protocol. To eliminate the single-stranded extensions, biotin-labeled DNA (0.1 μg/μl) was suspended in 1X Mung Bean nuclease buffer with 1U of Mung Bean Nuclease (NEB®,M0250S) per μg DNA at 30°C for 30 minutes.
PCR Selective Suppression Hybridization (PSSH)
Figure 1 shows the schematic diagram on principle of PCR Selective Suppression Hybridization (PSSH). The methylated and unmethylated DNA from NSCs was ligated with Ad1 and Ad2 adapters, respectively. Adapter sequences were adapted from Diatchenko et al. [9] and were custom-synthesized by Genomics® BioSci& Tech, Taiwan. The adapter sequences used are as follows:
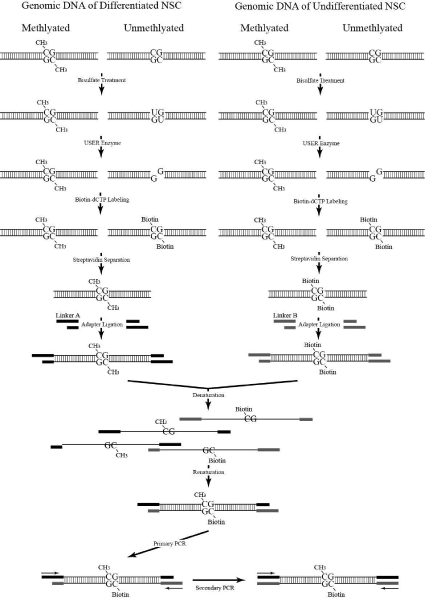
Figure 1: The Schematic Diagram of PSSH.
Ad1 (for methylated DNA):
5 ’ C T A A T A C G A C T C A C T A T A G G G C T C G A G C G GCCGCCCGGGCAGGT 3’
3’ GGCCCGTCCA 5’
Ad2 (for unmethylated DNA):
5’ C T A A T A C G A C T C A C T A T A G G G C A G C G T G G T C G C G G C C G A G G T 3’
3’ GCCGGCTCCA 5’
Separate ligation mixtures of 40 μL total volume containing a mixture of 1~2 μg DNA samples, 4 μL annealed adapters, 2 μL of 100 mM ATP (TAKARA, 4021), and 350 U of Ligase (TAKARA, 2011A) were incubated at 16°C for 16 hours in a cool block bath (SKYLINE, SKC40).
The adapter-ligated methylated and unmethylated samples (~200ng) were combined and mixed together in a 1.5mL eppendorf. The mixture was incubated in boiling water for 10 minutes and allowed to anneal by gradual cooling at room temperature. The primary PCR was conducted in a 25μL reaction mixture containing 5μL (approx. 100 ng) hybridized DNA, 1μL primer P1 (5’-CTAATACGACTC ACTATAGGGC-3’), 0.5μL dNTP (2.5 mM) and 5U GeneTAQ DNA polymerase suspended in proper PCR buffer and placed in a 200μL PCR tube. The mixture was centrifuged briefly prior to running in the thermal cycle machine (ABITM, GENAMP 9700) using the following parameters: 75°C for 5 minutes, 35 cycles at (95°C, 1 minute, 53°C, 1 minute, 72°C, 1 minute), and a final 7-minute extension step. A 5μL aliquot of the primary PCR product was used for the secondary PCR.
Methylated DNA from differentiated NSC was hybridized with the biotin-labeled DNA from the undifferentiated NSCs to determine the differentially methylated genes.
Secondary PCR was conducted similar to the primary PCR except for the primer, which was replaced with a mixture of the nested primers, NP1 (5’-TCGAGCGGCCGC CCGGGCAGGT-3’) and NP2 (5’-AGCGTGGTCGCGGCCGAGGT-3’). Parameters for the PCR run were also changed into the following: 95°C for 5 minutes, 25 cycles at (95°C, 1 minute, 61°C, 1 minute, and 72°C, 1 minute) plus a final 7-minute extension step. The PCR products were analyzed by 2% agarose gel electrophoresis and ethidium bromide staining (Figure 2).
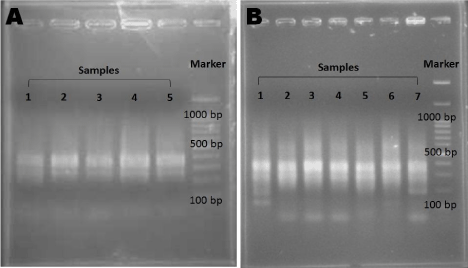
Figure 2: Validation of PCR Products.
The PCR products for (A) Primary PCR and (B) Secondary PCR were checked using 2% agarose gel electrophoresis.
TA cloning of PCR products
After successfully producing smear PCR products after secondary PCR, this process was used for selective sequence library. Obtained DNA fragments (3 μL of PCR product) were inserted in 1 μL of pGEM®-T Easy Vector (Promega) using 1 μL of T4 Ligase in 5 μL of 2X Rapid Ligation Buffer. Solutions were incubated at 14°C for 24 hours or longer. Briefly, at least 1μl of ligated TA cloning product was added to thawed 35-40μl electro competent DH5a cells incubated on ice for 30~60 seconds. One pulse was applied at 25μF, 2.1kV and 200O, giving a time constant of 4.5 to 5.0 milliseconds. After a short electric pulse, the cuvette was removed as quickly as possible and 1ml SOC medium was transferred into it. The cells were gently mixed and incubated at 37°C for 1 hour. Electroporated cells were then spread onto pre-warmed LB plates with ampicillin, IPTG, and X-Gal. The plates were incubated at 37°C for 12~16 hours. White transformed colonies were selected for cloned insert checking.
Picked colonies were checked by mixing with 1.2μL primer T7 and SP6 (3.0 μM), 0.5 μL dNTP (2.5 mM), and 5U of GeneTAQ DNA polymerase suspended in proper PCR buffer. The thermal cycler (ABITM, GENAMP 9700) ran under the following parameters: 95°C for 5 minutes, 35 cycles at (95°C, 1 minute, 62°C, 1 minute, 72°C, 1 minute), and a final 7 minute extension step. PCR products were run on 2% agarose gel and stained with ethidium bromide (EtBr) for visualization. The selected colonies with correct inserts were sent to Genomics® BioSci& Tech, Taiwan for sequencing. The successfully cloned and sequenced genes were aligned using Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) from the National Institutes of Health (NIH) to determine candidate genes. Candidate genes were also analyzed using DAVID (https://david.abcc. ncifcrf.gov/) and Gestalt (https://bioinfo.vanederbilt.edu/webgestalt/) which allowed these genes to be clustered based on function, and different gene ontologies. Moreover, Pathway Interaction Database (PID) (https://pid.nci.nih.gov/) defined them on the basis of their pathway interactions.
miRNA microarray data analysis
Cells cultured in POMA and PDL was maintained at a cell density range of 106-107. The culture was washed with 1X PBS. Then, the cells were treated with trypsin to detach the cells from the material. The enzymatic activity of trypsin was deactivated by adding an equal amount of media. The cells were collected by centrifuging at 1000 rpm for 5 minutes in room temperature and their total RNAs were extracted using TriZol. The same density of neurosphere was collected for total RNA extraction. The samples were sent to Phalanx Biotech Group (Hsinchu, Taiwan) for microarray analysis. The standard selection criteria for differentially expressed transcripts were with statistical significance taken at log2 ratio of =±0.8 and/or P-value <0.05 for miRNAs.
Normalized intensity was obtained by normalizing with 75% scaling normalization method, then take average of repeated data from the same sample. CV is the correlation coefficient of the repeated probes within one chip. Normalized spot intensities were transformed to gene expression log2 ratios. 1.74 Fold-Change means log2 ratios >= 0.8 or log2 ratios =< -0.8. P-value of differentially expressed transcripts is calculated with two samples t-test. It means the probability of control sample and test sample are no differential expression. So, if p-value (Differentially expressed) is greater than the threshold it means no differential repression was observed.
Results and Discussion
Differentially methylated genes involved in NSC differentiation on electrospun POMA fibers
Neural stem cells were seeded on electrospun POMA fibers to 10cm petri dishes and allowed to differentiate for 7 days. Addition of the drug dbcAMP was omitted due to its negative effect on cell viability after 5 days in culture, especially at high initial cell seeding densities (7x105 and 1x106). POMA was observed to facilitate efficient cell attachment, promote and support rapid cell proliferation, and enable differentiation into predominantly neuron precursor cells in culture without drug, while cells treated with dbcAMP exhibited more glial cell star-shaped morphology (Figure 3).
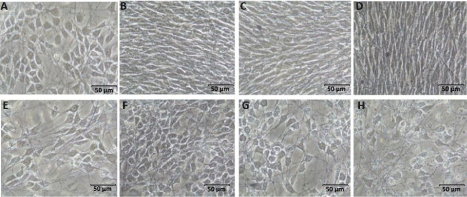
Figure 3: NSCs cultured on POMA without dbcAMP for (A) 1 day (B) 3 days (C) 5 days (D) 7 days and with dbcAMP (E-H).
Genomic DNA from rat neural stem cells in primary culture and from differentiated NSCs on biomaterials were extracted and screened for differentially methylated genes involved in rNSCs differentiation. Differentially methylated DNA was obtained through PSSH technique. DNA fragments cloned in E. coli belonged to cells differentiated on POMA in the presence of dbcAMP. The gene identity of the cloned inserts was determined using BLAST. These genes were further analyzed using three different bioinformatics databases that allowed them to be clusetered based on function (DAVID and Gestalt), different gene ontologies (Gestalt and PID), and delineated on the basis of their Pathway Interactions (PID).
Among the 69 inserts sequenced, 17 genes were confirmed to be actively involved in differentiation (Table 1). There are also genes that influence stem cell fate without direct interaction with the inserts. The protein products of these genes usually act as transcription factors (activators, enhancers, repressors) that regulate the expression of
![]()
Full Name
Symbol and synonyms
Hyperpolarization-activated cyclic nucleotide-gated potassium channel 1
HCN1
Coagulation factor II (thrombin) receptor
F2R, PAR-1
Engulfment and cell motility 1
ELMO1
POU class 3 homeobox 3
POU3F3, BRN1, OTF8
Diacylglycerol lipase, alpha
DAGLA
Dystrophin
DMD
Cdk5 and Abl enzyme substrate 1
CABLES1
Heat shock protein B8
HSPB8
Kinesin family member 5C
KIF5C, Kinesin 1C
Histone deacetylase 8
HDAC8
Neural cell adhesion molecule 2
NCAM2
Retinoblastoma 1
RB1
RAB38, member RAS oncogene family
RAB38
chemokine-like receptor 1
CMKLR1
Fizzy/cell division cycle 20 related 1 (Drosophila)
FZR1, EMI1
Glutamate receptor, ionotropic, delta 1
GRID1
Zinc finger E-box binding homeobox 1
ZEB1
Table 1: Sequenced genes directly associated with stem cell proliferation and differentiation.
cell-specific genes. Candidate genes with such function, as reported in literature, and which were extracted from the sample grown on a biomaterial (POMA), are listed in Table 2.
![]()
Full Name
Symbol and synonyms
Related gene(s) or pathway
T-cell lymphoma invasion and metastasis 1
TIAM1
RAC1
M-phase phosphoprotein 1, also known as Kinesin family member 20b
MPP1, KIF20B, KRMP1
PIN1, Nanog
Deleted in colorectal carcinoma (netrin receptor)
DCC
Netrin, MAPK
eukaryotic translation initiation factor 5B
EIF5B
Mei-P26
solute carrier organic anion transporter family, member 4C1
SLCO4C1
SOX17
T-cell leukemia/lymphoma 1A
TCLLA
AKT1, N-CoR
potassium inwardly-rectifying channel, subfamily J, member 6
KCNJ6, GIRK2
Bcl-XL
cell division cycle 73, Paf1/RNA polymerase II complex component, homolog (S. cerevisiae)
CDC73, HRPT2, parafibromin
WNT,
Wnt signaling pathway
FAT tumor suppressor homolog 4 (Drosophila)
FAT4, CDHF14
Hippo pathway, EGFR/Ras signaling pathway
Table 2: Candidate genes indirectly involved in stem cell differentiation.
For the confirmed genes, KIF5C and NCAM2 were found to have roles in cell differentiation and neuron projection morphogenesis.
KIF5C or kinesin family member 5C encodes a protein that, in mammals, serves as part of the assembly of Kinesin-1, a major anterograde motor that drives transport into the axons of neurons [11]. In addition, the kinesin light chain 1 (KLC1) or kinesin-1 subunit is said to be essential for normal human neural differentiation [12].
NCAM2, or the neuronal adhesion cell molecule 2, belongs to the Immunoglobulin (Ig) super family. NCAM2 contains consensus sequences for posttranslational modifications and is involved in several neurological disorders such as autism [13]. NCAM2 also has functions in neural stem cell adhesion and is expressed early on in differentiation but later on repressed [14]. Another confirmed gene that DAVID clustered in cell differentiation and neuron projection morphogenesis and Gestalt grouped under cell differentiation is DMD or dystrophin, muscular dystrophy gene. DMD is a member of the Dystrophin/Dystroglycan Glycoprotein (DGC) complex that is stabilized by the Lis1-Nde1 complex responsible for maintaining radial glial cell functions [15]. Radial glial cells are distinctive, bipolar stem cells found in the cerebral cortex.
Another confirmed gene related by PID to differentiation is the coagulation factor II or thrombin receptor F2R, a blood coagulation factor found to be differentially expressed in neural, embryonic, and hematopoietic stem cells, but not in adult cells [16-18]. F2R shares a similar pathway with the gene GRID1, a glutamate receptor required for neuronal function, ion transport, and cell motility. GRID1 is silent in stem cells but only H3K27 trimethylated in progenitor cells, and it is induced upon terminal differentiation, losing its trimethylation at lysine 27 of histone 3 [19]. According to Gestalt, GRID1 and F2R are part of the neuroactive ligand-receptor interaction pathway necessary for environmental information processing.
Genes that are connected or are actively involved in the same pathways are listed accordingly in Table 3. Based on Pathway Interaction Database (PID) analysis, confirmed genes and those that have indirect interactions with differentiation genes have a distant relationship with each other within the pathways they are included in. Only ELMO1 and TIAM1 have established relationships with the other genes, hinting of a broader influence and a bigger impact on the process of differentiation.
![]()
Confirmed genes
Candidate genes
Pathway
Database
ELMO1
DCC
Netrin-mediated signaling events
PID
ELMO1
TIAM1
Neurotrophic factor-mediated Trk receptor signaling,
PID
Chemokine signaling pathway
Gestalt
ELMO1
RASGRF2, TIAM1, VAV1, VAV2
Regulation of RAC1 activity
PID
CDH1
TIAM1, VAV2
E-cadherin signaling in the nascent adherens junction, CDC42 signaling events
PID
CABLES1, CDH1
TIAM1
Pos-translational regulation of adherens junction stability and disassembly
PID
CDH1
VAV2
Nectin adhesion pathway
PID
FZR1
ECT2
PLK1 signaling events
PID
FZR1
RB1
Cell cycle
Gestalt
F2R
TIAM1, MON1B, VAV1,
Regulation of actin cytoskeleton
Gestalt
F2R/PAR
GRID1
-
Neuroactive ligand-receptor interaction
Gestalt
RB1
DCC
Pathways in cancer
Gestalt
Table 3: Relationship between confirmed genes and candidate genes.
Aside from taking part in the neurotrophic factor-mediated Trk receptor signaling (PID), they also appear in the chemokine signaling pathway responsible for actin cytoskeleton regulation (Gestalt). The neurotrophic factor-mediated Trk receptor signaling mediates transcription factors (C/EBPa, -ß, and neuroD) recruitment to early gene promoters in order to induce immediate-early gene expression [20]. Another pathway TIAM1 and ELMO1 share along with RASGRF2, VAV1, and VAV2 is the regulation of RAC1 activity. Loss of RAC1 expression results in reduced proliferation and subsequent differentiation [21].
ELMO1 is also involved in the netrin-mediated signaling events along with DCC, an axon guidance receptor that responds to Netrin-1. Netrin-1 activates the MAPK pathway in order to guide growth cones. MAPK, in turn, directly interacts with DCC, suggesting that netrin is involved in the proliferation or differentiation of neural precursor cells [22]. DCC is also associated with pathways in cancer along with RB1, a known tumor suppressor that triggers cell cycle arrest which, in turn, mediates cell-to-cell contact inhibition [23]. RB1 is also an important regulator of cell proliferation and differentiation [24]. Gestalt confirmed RB1’s presence in the cell cycle pathway along with FZR1. FZR1 is similar to Xenopus Early Mitotic Inhibitor (EMI1) hence it inhibit the activity of the anaphase-promoting complex/cyclosome (APC/C) during S phase until the prophase. It was found that depletion of EMI1 results in loss of stem cell identity and trophoblast differentiation in embryonic stem cells [25].
TIAM1 is also involved in 3 other pathways, namely: the E-cadherin signaling in the nascent adherens junction along with CDH1 (FZR1), in post-translational regulation of adherens junction stability and disassembly with both CDH1 and CABLES1, and in actin cytoskeleton regulation with F2R. CABLES 1 is a cyclin-dependent kinase-interacting protein known to inhibit cell cycle progression [26]. It is also an important protein for early neural differentiation [27].
HCN1 is another confirmed gene whose function is also required in neuron projection morphogenesis (DAVID). HCN1 belongs to a group of ion channels significant for sensory transduction and cellular development. Hyperpolarization-activated cyclic nucleotide-gated channels have also been found to exert pro-proliferation effects in embryonic stem cells [28]. They also carry a particular current I(h) that resembles the current found in neurons and cardiomyocytes, as well as mediate the generation of pacemaker activity in the heart and brain [29]. POU3F3 is a transcription factor that binds to the enhancer of the rat nestin (intermediate filament protein) gene and the structurally similar regulatory site of the Brain Fatty Acid Binding Protein (B-FABP), both of whom are neural stem cell markers, to activate expression of neural stem cell-specific properties in cooperation with hormone response elements [30]. DAGLA or diacylglycerol lipase, alpha functions in axonal growth and guidance during development, retrograde synaptic signaling at mature synapses, and maintenance of adult neurogenesis. Expression of DAGLA is down-regulated when neural stem cells are differentiated into neurons [31]. HSPB8 is a member of the small heat shock family of proteins that is greatly expressed in the cerebellum, cortex, heart, and muscle, as well as in motor and sensory neurons. It is also involved in cytoprotection via apoptosis blocking and is associated in cell metabolic activities. HSPB8 is highly expressed during differentiation [32]. HDAC8 is a ubiquitously expressed Class I deacetylase that can induce differentiation and cell cycle arrest, inhibit clonogenic growth, and reduce the proliferation of neuroblastomas upon knockdown [33]. RAB38 belongs to a family of small GTPases involved in intracellular signaling processes like tethering and docking of vesicles to their target compartment, vesicle budding, etc. It is also an identified stem cell marker [34]. Finally, CMKLR1 is a cognate receptor that plays a role in adipocyte and osteoblast differentiation [35].
All of these identified sequences are considered unmethylated and functional before differentiation of NSCs but were silenced by methylation after differentiation. Of the 17 confirmed genes which are involved in NSC lineage commitment, eleven genes corresponding to neural stem cell functions were appropriately methylated, indicating that the POMA scaffold was efficient in enhancing differentiation of NSCs with the help of dbcAMP. These indicators of proper differentiation are: NCAM2, ELMO1, KIF5C,DMD, F2R, FZR1, HCN1, DAGLA, HSPB8, RAB38, and CMKLR1, whose respective expressions should be turned off once NSCs differentiate.
miRNA regulation during NSC differentiation on POMA
The expression of miRNAs on PDL and POMA was normalized on the basis of the miRNA expression of undifferentiated NSCs. The differentially expressed transcripts were selected with a criteria of having |log2 ratio | = 0.8 and P-value < 0.05. Differentially expressed miRNAs during NSC differentiation are shown in Table 4 (POMA) and 5 (PDL).
![]()
Name
Normalized Intensity
CV
log2 (Ratio)
P-value
NSC
POMA
NSC
POMA
POMA/NSC
POMA/NSC
Upregulated
rno-miR-1224
22260.5000
42875.9569
6.89E-02
0.00E+00
0.9457
2.76E-03
rno-miR-204-3p
7280.1151
16412.1084
0.00E+00
0.00E+00
1.1727
0.00E+00
rno-miR-25-5p
411.9822
726.8069
2.15E-02
6.29E-02
0.8190
1.07E-02
rno-miR-30c-1-3p
2413.2447
6881.2467
2.93E-02
0.00E+00
1.5117
1.25E-04
rno-miR-92a-2-5p
3454.7377
6160.4964
1.80E-01
0.00E+00
0.8345
2.55E-02
rno-miR-92b-5p
5543.7327
10315.6114
0.00E+00
8.90E-02
0.8959
1.80E-02
Downregulated
rno-miR-208a-5p
625.9053
253.1711
1.41E-01
3.16E-02
-1.3058
2.72E-02
rno-miR-221-3p
344.1998
144.3519
5.33E-02
3.76E-02
-1.2537
4.56E-03
rno-miR-222-3p
140.1316
78.7374
2.67E-02
0.00E+00
-0.8317
1.85E-03
rno-miR-347
341.0751
162.3202
1.09E-01
1.51E-01
-1.0712
2.96E-02
rno-miR-375-5p
440.8258
124.3647
1.47E-01
0.00E+00
-1.8256
2.02E-02
Table 4: Microarray data of the differentially miRNAs during NSC differentiation on POMA.
![]()
Name
Normalized Intensity
CV
log2 (Ratio)
P-value
PDL
NSC
PDL
NSC
PDL/NSC
PDL/NSC
Upregulated
rno-miR-1-3p
206.0705
956.1549
5.30E-02
1.73E-02
2.2141
3.50E-04
rno-miR-105
120.4224
288.5020
3.03E-02
6.63E-02
1.2605
6.64E-03
rno-miR-1188-5p
322.6291
660.1830
1.27E-01
1.15E-01
1.0330
3.12E-02
rno-miR-122-5p
167.4323
515.6292
2.72E-02
0.00E+00
1.6228
8.55E-05
rno-miR-125b-1-3p
200.9187
351.8957
0.00E+00
9.98E-02
0.8085
2.60E-02
rno-miR-187-3p
153.2649
504.9289
4.75E-02
8.20E-02
1.7201
7.07E-03
rno-miR-206-3p
211.2222
755.8792
1.72E-02
1.27E-01
1.8394
1.52E-02
rno-miR-210-3p
291.7185
1175.8121
6.87E-02
8.21E-02
2.0110
6.16E-03
rno-miR-214-5p
159.3826
544.0958
2.57E-02
1.62E-01
1.7714
2.54E-02
rno-miR-297
522.5819
2686.7630
6.36E-02
1.20E-01
2.3621
1.10E-02
rno-miR-32-3p
598.5703
4483.7932
2.81E-02
9.80E-02
2.9051
6.34E-03
rno-miR-330-5p
128.1501
270.5337
7.11E-03
3.59E-02
1.0780
2.34E-03
rno-miR-346
371.8928
678.7569
1.10E-02
3.28E-02
0.8680
2.71E-03
rno-miR-3557-3p
189.9712
483.3266
4.79E-03
8.27E-03
1.3472
9.76E-05
rno-miR-3562
622.0753
1251.7231
1.90E-02
8.17E-02
1.0088
1.31E-02
rno-miR-3594-5p
251.1484
463.9451
1.45E-02
5.05E-02
0.8854
6.14E-03
rno-miR-382-5p
113.9827
215.0137
2.40E-02
7.57E-02
0.9156
1.31E-02
rno-miR-410-5p
124.9302
260.0354
2.92E-02
3.29E-02
1.0576
2.36E-03
rno-miR-465-5p
364.1652
1417.6773
3.75E-03
0.00E+00
1.9609
8.41E-07
rno-miR-466b-5p
3690.2715
20171.3155
4.16E-02
0.00E+00
2.4505
4.33E-05
rno-miR-466c-5p
325.2050
1910.6947
8.40E-03
9.18E-02
2.5547
6.06E-03
rno-miR-466d
1518.8038
10394.9545
7.95E-02
9.92E-02
2.7749
6.77E-03
rno-miR-494-5p
89.1899
158.4843
1.53E-02
3.06E-02
0.8294
2.64E-03
rno-miR-496-5p
109.7969
267.3034
5.39E-02
0.00E+00
1.2836
7.06E-04
rno-miR-505-5p
194.8010
540.8655
7.01E-03
1.58E-03
1.4733
1.09E-05
rno-miR-672-5p
1221.2896
7940.7697
4.74E-02
0.00E+00
2.7009
3.70E-05
rno-miR-760-3p
367.7070
674.9210
2.48E-03
8.16E-02
0.8762
1.57E-02
rno-miR-92a-2-5p
2593.9121
6160.4964
3.93E-02
0.00E+00
1.2479
4.09E-04
rno-miR-99b-3p
217.6619
905.6822
8.37E-03
3.22E-02
2.0569
8.98E-04
Downregulated
rno-miR-122-3p
270.7895
125.1723
2.19E-02
9.58E-02
-1.1133
4.19E-03
rno-miR-196c-3p
236.9811
110.4343
6.92E-02
1.29E-02
-1.1016
8.35E-03
rno-miR-208a-5p
872.5797
253.1711
0.00E+00
3.16E-02
-1.7852
8.33E-05
rno-miR-290
712.8751
241.8652
0.00E+00
1.42E-01
-1.5594
2.64E-03
rno-miR-292-5p
249.5385
117.5005
5.66E-02
8.26E-02
-1.0866
8.31E-03
rno-miR-296-5p
245.6746
96.9076
2.41E-02
5.89E-03
-1.3421
7.98E-04
rno-miR-29b-3p
248.5725
115.4815
4.03E-02
6.43E-02
-1.1060
4.36E-03
rno-miR-347
439.1877
162.3202
1.24E-02
1.51E-01
-1.4360
4.10E-03
rno-miR-3574
156.4848
88.2263
0.00E+00
2.27E-02
-0.8267
4.28E-04
rno-miR-3593-3p
358.3694
168.9826
2.67E-02
7.27E-02
-1.0846
3.36E-03
rno-miR-3596a
265.3157
84.5923
4.81E-02
4.39E-02
-1.6491
2.69E-03
rno-miR-3596c
1645.3440
520.2726
9.52E-02
5.43E-02
-1.6610
9.86E-03
rno-miR-375-5p
647.1901
124.3647
1.97E-02
0.00E+00
-2.3796
2.97E-04
rno-miR-466b-1-3p
1176.5336
194.0171
4.64E-03
1.19E-01
-2.6003
2.92E-04
rno-miR-466b-2-3p
263.3838
68.0372
1.73E-02
5.46E-02
-1.9528
4.52E-04
rno-miR-466c-3p
467.5224
135.4687
4.29E-02
8.22E-02
-1.7871
2.37E-03
rno-miR-615
612.0937
226.7234
3.72E-03
3.15E-02
-1.4328
1.89E-04
Table 5: Microarray data of the differentially miRNAs during NSC differentiation on PDL.
IPA clustered the differentially regulated miRNAs based on their seed sequences and identified the highly predicted and experimentally observed target mRNAs (Table 4 and 5). Target mRNAs were gathered from all identified differentially expressed miRNAs (upregulated and downregulated) from both substrates. The target mRNAs from differentially miRNAs from both substrates were obtained using IPA and miRbase.
POMA up-regulated miR-1224, miR-204-3p, miR-30c-1-3p, and miR-92-5p. The miR-1224 had been reported as a potential marker for acetaminophen which effects central nervous system to relieve pain, induced liver injury [36]. miR-204 was found to have a putative function in neural differentiation processes, including axon guidance as supported by in vivo functional studies [37]. Knockdown of miR- 204 resulted to axon path finding defects.miR-25 and miR-30c are upregulated after NSC differentiation on POMA. These miRNAs regulate the proliferation and differentiation of neural adult and progenitor stem cells. Knocking down miR-25 decreases NSPC proliferation, whereas ectopically expressing miR-25 promotes NSPC proliferation. Expression of miR-25 in NSPCs increases their ability to generate new neurons. On the other hand, elevated miR- 30c increased neuronal proliferation in the SVZ of mouse [38]. Also, miR-30c participates in expression of p53 gene which may associated with cell apoptosis [39]. Lastly, miR-92 has a significant upregulated expression on both POMA and PDL. A study showed that miR- 92a and miR-92b maintain neuroblastself-renewal by inhibiting premature differentiation in Drosophilia [40]. The miR-92 family has a negative feedback loop as an essential regulator in neural stem cell development. The expression of these miRNAs further enhanced the differentiation and proliferation potential of POMA as a scaffold for NSC differentiation.
Conclusion
In this study, a neat electrospun poly-o-methoxyaniline (POMA) was synthesized and its efficiency as a substrate for NSC differentiation were evaluated. It was found that of the seventeen confirmed genes which are involved in NSC lineage commitment, eleven genes corresponding to neural stem cell functions were appropriately methylated, indicating that the POMA scaffold was efficient in enhancing differentiation of NSCs with the help of dbcAMP. These indicators of proper differentiation are: NCAM2, ELMO1, DMD, F2R, FZR1, HCN1, DAGLA, HSPB8, RAB38, and CMKLR1, whose respective expressions should be turned off once NSCs differentiate. Furthermore, the miRNA microarray data indicated that the expression of miR-1224, miR-204-3p, miR-30c-1-3p, and miR-92-5p were differentially upregulated as compared to PDL.The expression of these miRNAs further enhanced the differentiation and proliferation potential of POMA as a scaffold for tissue engineering.
Acknowledgement
The authors gratefully acknowledge the National Science Council of Taiwan ROC (NSC-102-2632-M-033-001-MY3; NSC99-2632-M- 033-001-MY3) and Chung Yuan Christian University, Taiwan ROC for supporting the research work.
References
- Cao Q, Benton RL, Whittemore SR. Stem cell repair of central nervous system injury. J Neurosci Res. 2002; 68: 501-510.
- Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005; 21: 605-631.
- Ravichandran R, Sundarrajan S, Venugopal JR, Mukherjee S, Ramakrishna S. Applications of conducting polymers and their issues in biomedical engineering. Journal of the Royal Society Interface. 2010; 7: 559-579.
- Li D, Huang JX, Kaner RB. Polyaniline Nanofibers: A Unique Polymer Nanostructure for Versatile Applications. Accounts of Chemical Research. 2009; 42: 135-145.
- Chung-Feng Dai, Chang-Jian Weng, Tzu-Chun Yeh, Bo-Cheng Lai, Chang-Yu Sung, Yen Wei, et al. Preparation of electrospun electroactive POMA fiber mats. Polymer International. 2012; 61: 213-221.
- Lu-Chen Yeh, Chung-Feng Dai, Jui-Ming Yeh, Ping-Yi Hsieh, Yen Wei, Ting-Yu Chin, et al. Neat poly (ortho-methoxyaniline) electrospun nanofibers for neural stem cell differentiation. Journal of Materials Chemistry B. 2013; 1: 5469-5477.
- Juliandi B, Abematsu M, Nakashima K. Epigenetic regulation in neural stem cell differentiation. Dev Growth Differ. 2010; 52: 493-504.
- Genereux DP, Johnson WC, Burden AF, Stöger R, Laird CD. Errors in the bisulfite conversion of DNA: Modulating inappropriate and failed-conversion frequencies. Nucleic Acids Res. 2008; 36: e150.
- Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci. USA. 1996. 93: 6025-6030.
- National Research Council (US). Committee for the Update of the Guide for the Care and Use of Laboratory Animals., Institute for Laboratory Animal Research (US), and National Academies Press (US), Guide for the care and use of laboratory animals. National Academies Press, Washington, D.C. p. 1 online resource. 2011; 220.
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N, et al. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci. 2000; 20: 6374-6384.
- Killian RL, Flippin JD, Herrera CM, Almenar-Queralt A, Goldstein LS. Kinesin light chain 1suppression impairs human embryonic stem cell neural differentiation and amyloid precursor protein metabolism. PLoS One. 2012; 7: e29755.
- Kulahin N, Walmod PS. The neural cell adhesion molecule NCAM2/OCAM/RNCAM, a close relative to NCAM. Adv Exp Med Biol. 2010; 663: 403-420.
- Wang CC, Kadota M, Nishigaki R, Kazuki Y, Shirayoshi Y, Rogers MS, et al. Molecular hierarchy in neurons differentiated from mouse ES cells containing a single human chromosome 21. Biochem Biophys Res Commun. 2004; 314: 335-350.
- Pawlisz AS, Feng Y. Three-dimensional regulation of radial glial functions by Lis1-Nde1 and dystrophin glycoprotein complexes. PLoS Biol. 2011; 9: e1001172.
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA M. "Stemness": Transcriptional profiling of embryonic and adult stem cells. Science. 2002; 298: 597-600.
- Ryser S, Glauser D, Vigier M, Zhang YQ, Tachini P, Schlegel W, et al. Gene expression profiling of rat spermatogonia and Sertoli cells reveals signaling pathways from stem cells to niche and testicular cancer cells to surrounding stroma. BMC Genomics. 2011; 12: 29.
- Sainz J, García-Alcalde F, Blanco A, Concha A. Genome-wide gene expression analysis in mouse embryonic stem cells. Int J Dev Biol. 2011; 55: 995-1006.
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008; 30: 755-766.
- Calella AM, Nerlov C, Lopez RG, Sciarretta C, von Bohlen und Halbach O, Bereshchenko O, et al. Neurotrophin/Trk receptor signaling mediates C/EBP alpha, -beta and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Development. 2007; 2:4.
- Leone DP, Srinivasan K, Cord Brakebusch, Susan K, McConnell. The Rho GTPase Rac1 is required for Proliferation and Survival of Progenitors in the Developing Forebrain. Developmental Neurobiology. 2010. 70: 659-678.
- Aoki M, Yamashita T, Tohyama M. EphA receptors direct the differentiation of mammalian neural precursor cells through a mitogen-activated protein kinase-dependent pathway. Journal of Biological Chemistry. 2004; 279: 32643-32650.
- Liu Y, Clem B, Zuba-Surma EK, El-Naggar S, Telang S, Jenson AB, et al. Mouse Fibroblasts Lacking RB1 Function Form Spheres and Undergo Reprogramming to a Cancer Stem Cell Phenotype. Cell Stem Cell. 2009; 4: 336-347.
- de Bont JM, Packer RJ, Michiels EM, den Boer ML, Rob Pieters. Biological background of pediatric medulloblastoma and ependymoma: A review from a translational research perspective. Neuro-Oncology. 2008; 10: 1040-1060.
- Yang VS, Carter SA, Hyland SJ, Tachibana-Konwalski K, Laskey RA, Gonzalez MA, et al. Geminin Escapes Degradation in G1 of Mouse Pluripotent Cells and Mediates the Expression of Oct4, Sox2, and Nanog. Current Biology. 2011; 21: 692-699.
- Lee HJ, Sakamoto H, Luo H, Skaznik-Wikiel ME, Friel AM, Niikura T, et al. Loss of CABLES1, a cyclin-dependent kinase-interacting protein that inhibits cell cycle progression, results in germline expansion at the expense of oocyte quality in adult female mice. Cell Cycle. 2007; 6: 2678-2684.
- Groeneweg JW, White YAR, David K, Peterson RT, Zukerberg LR, Inna Berin, et al. cables1 is required for embryonic neural development: Molecular, cellular, and behavioral evidence from the Zebrafish. Molecular Reproduction and Development. 2011; 78: 22-32.
- Jehle J, Schweizer PA, Katus HA, Thomas D. Novel roles for hERG K (+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011; 2: e193.
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 1997; 94: 14815-14820.
- Josephson R, Müller T, Pickel J, Okabe S, Reynolds K, Turner PA, et al. POU transcription factors control expression of CNS stem cell-specific genes. Development. 1998; 125: 3087-3100.
- Walker DJ, Suetterlin P, Reisenberg M, Williams G, Doherty P. Down-regulation of diacylglycerol lipase-alpha during neural stem cell differentiation: identification of elements that regulate transcription. J Neurosci Res. 2010; 88: 735-745.
- Choi MR, Jung KH, Park JH, Das ND, Chung MK, Choi IG, et al. Ethanol-induced small heat shock protein genes in the differentiation of mouse embryonic neural stem cells. Arch Toxicol. 2011; 85: 293-304.
- Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Letters. 2009; 277: 8-21.
- Krzyzanowski PM, Andrade-Navarro MA. Identification of novel stem cell markers using gap analysis of gene expression data. Genome Biology. 2007; 8.
- Muruganandan S, Roman AA, Sinal CJ. Role of Chemerin/CMKLR1 Signaling in Adipogenesis and Osteoblastogenesis of Bone Marrow Stem Cells. Journal of Bone and Mineral Research. 2010; 25: 222-234.
- Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci USA. 2009; 106: 4402-4407.
- Conte I, Merella S, Garcia-Manteiga JM, Migliore C, Lazarevic D, Carrella S, et al. The combination of transcriptomics and informatics identifies pathways targeted by miR-204 during neurogenesis and axon guidance. Nucleic Acids Res. 2014; 42: 7793-7806.
- Sun T, Li W, Ling S. miR-30c and semaphorin 3A determine adult neurogenesis by regulating proliferation and differentiation of stem cells in the subventricular zones of mouse. Cell Prolif. 2016; 49: 270-280.
- Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010; 6: e1000795.
- Yuva-Aydemir Y, Xu XL, Aydemir O, Gascon E, Sayin S, Zhou W, et al. Downregulation of the Host Gene jigr1 by miR-92 Is Essential for Neuroblast Self-Renewal in Drosophila. PLoS Genet. 2015; 11: e1005264.