
Research Article
Austin J Biotechnol Bioeng. 2014;1(1): 8.
In-silico Approach to Map Transcription Factor Binding Motifs onto Drosophila Cardiac Genes
Jain Prerna1 and Hasija Yasha1*
1Department of Biotechnology, Delhi Technological University, India
*Corresponding author: Mahendra Rai K, Department of Biotechnology, Sant Gadge Baba Amravati University, Amravati- 444602 Maharashtra, India.
Received: June 30, 2014; Accepted: July 25, 2014; Published: July 28, 2014
Abstract
The development of multi cellular organisms requires a consortium of different types of cells that interact to form a functional organism. Each cell has a unique genetic profile that is regulated in a specific manner according to different developmental stages of an organism. This control is orchestrated by regulatory sequences called enhancers which are gene regulatory sequences that dictate the spatio-temporal patterns of gene expression by controlling transcriptional activities. A common feature of the regulatory enhancers is the presence of multiple binding sites known as Transcription Factor Binding Sites (TFBS), which binds to multiple transcription factors. A molecular understanding of enhancers and various transcription factors that bind to these is necessary for determining complex biological networks. The binding sites within the enhancers are conserved in nature, thus finding out those sites can help in uncovering various interaction mechanisms. In the present study, we try to address this issue by computational approach to predict TFBS within the set of enhancers in Drosophila melanogaster heart organ. We collected all the known enhancers that are active in cardiac mesoderm. The motifs were identified and functionally characterized by comparing with the database of known motifs. Putative motifs were mapped onto our dataset of enhancer sequences. We believe that these mapped enhancer sequences can be used to predict various de novo enhancers in the entire Drosophila melanogaster genome using machine learning techniques. Thus these findings helps to discover mechanisms currently unknown and may be important in gene regulation.
Keywords: Transcription factor binding sites, Enhancers, Drosophila melanogaster, Cis regulatory module
Abbreviations
18w: 18 wheeler; Act 57b: Actin 57b; Atet: ABC Transporter Expressed in Trache; Bib: Big Brain; CRM: Cis Regulatory Module; Hh: Hedgehog; Kb: kilobase; Lea: Leak; MAST: Motif Alignment and Scan Tool; MEME: Multiple Em For Motif Elicitation; Mef2: Myocyte Enhancer Factor; Nkd: Naked cuticle; RC: Reporter Construct; Slp: Sloppy paired 1; Sur: Schmalspur; Tin: Tinman; Tl: Toll; TF: Transcription Factor; TFBS: Transcription Factor Binding Site
Introduction
Gene expression and control
Embryonic development is a tightly controlled process that ultimately leads to the development of a multi cellular organism comprising of complex tissues and organs. The development of different organs largely relies on the differentiation process, cell specification, cellular identity, as well as responses to environmental cues [1]. The precise spatio-temporal control of gene expression is the main driving force to the proper restriction of cell fates and for insuring the accuracy of cellular differentiation. This results in time-dependent and tissue-specific regulatory outputs, which are critical in regulating different stages of embryonic development. The knowledge of these transcriptional activation states at the right stage and time depends on several factors including the position of the gene in the genome, its chromatin structure and the transcriptional regulatory elements associated with each gene. These transcriptional regulatory elements play a major role in regulating gene expression and further decide the cell fate of the various cells in the developmental process [1,2].
During transcription, various transcription factors (TF) are involved, that bind to DNA in a specific sequence manner. The TFs bind to sequences called as transcription factor binding sites (TFBS) in the regulatory regions of the gene called enhancers which are organized in the form of modules, called as Cis-Regulatory Module (CRM). CRM sare regulatory sequences located few kilo bases away from gene of interest and bind to specific TFs at specific developmental stage to result in specific cell specification [3]. Overall, gene expression is regulated by the combination of all CRMs acting on genes throughout the organism's life. Previous studies have shown that gene encoding Transcription Factor tinman, has 4 CRMs controlling its expression which is a consequence of genetic pleiotropy. Thus, there exists as many as 10-fold more CRMs than genes [4].
Interaction between the TFs and CRMs form a development transcriptional regulatory network, encoding the specification and differentiation programmes of various cell types that are expressed at a particular stage in the development and finally lead to a full grown organism.
The prediction of these regulatory motifs, TFBS form an essential link in comparative genomics. These sequences are evolutionary conserved, and eventually we can find out the orthologous of these genes in higher and complex organism which help in understanding molecular mechanisms. But some of the hurdles to predictions are: these modules are located far away from the genes they regulate. Next, the presence of multiple transcription factor binding sites for various TFs leads to combinatorial control of gene regulation, thus making it difficult to associate with one gene [5,6]. The traditional approach to prediction involves the use of whole genome and techniques such as chromatin immune precipitation (ChIP) and ChIP-Seq to test many sequence fragments for regulatory activity in a reporter gene assay. These assays are highly intensive and these cannot assay all the tissues under all conditions [7]. Thus computational tools have been used to predict all the modules and binding sites effectively for example, one strategy is to scan whole genome in search of certain sequence based signatures which can be TFBS or specific histone modification based signatures. The data and the signatures are curated from published literature. These predictions can be uncertain, thus further experimental validations are always necessary [5-7].
Overview of Drosophila melanogaster Heart Development
Drosophila melanogaster as a model organism
Drosophila melanogaster is one of the most intensively studied organisms in biology and serves as a model system for the investigation of many developmental and cellular processes common to higher eukaryotes, including humans, thus an ideal system for developing and evaluating comparative genomics methodologies [8]. The annotated genome sequence of D. melanogaster, together with its associated biology, helps in unravelling various cellular and metabolic mechanisms. The first organ to be formed during embryogenesis is heart and is necessary to circulate blood systemically and support the progression of organogenesis. The early events in Drosophila heart development have been studied in detail. Previous studies have provided information about various factors involved in the early determination and differentiation of the cardiac mesoderm. Some of the factors such as Tin are evolutionary conserved and their homologues have been identified [9].
Studies have shown that large proportion of the diversity of living organisms results from differential regulation of gene transcription. Transcriptional regulation between species differs due to changes in interaction of TFs with enhancer sequences. These changes are very important criterion to specify which gene is expressed and at which stage. These mechanisms by which protein: DNA interactions evolve are therefore an important question in evolutionary biology [10,13]. Present work involves identifying and analyzing the genes that are specifically expressed in drosophila heart cells.
Cardiac specific genes and transcription factors
Cardio genesis proceeds via the activation of a complex regulatory network of cardiac structural genes. Significant progress has been made in defining the genes which contribute to heart development, in previous studies [11]. Figure 1 represents participation of various genes in the development of cardiac, visceral, skeletal muscles from dorsal mesoderm.
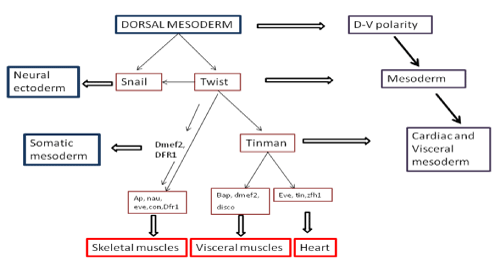
Figure 1: Various genes participating in the development of cardiac, visceral, skeletal muscles from dorsal mesoderm.
Genes That Affect Heart Development in Drosophila melanogaster
The important genes involved in the cardiac cell restriction are tinman and bagpipe. Tinman becomes restricted to visceral mesoderm first and in later stages to the cardiac mesoderm. In heart, tinman is expressed continuously. On one hand, where tinman is essential for cardiac development, on the other hand gene bagpipe has null effect on heart but major effect on visceral mesoderm, where a mutation in bagpipe deletes a large portion of visceral mesoderm [12].
Tinman
Transcription factor Tinman activates a number of key regulatory genes which mediate heart development thus, is critical to cardiac specification. Most of the cardiac enhancers for these Tin target genes contain at least two Tin binding sites, which are critical to normal cardiac function. The ability to easily and successfully identify cardiac enhancers based upon the presence of the Tin sites stands to greatly enhance our understanding of transcriptional regulatory networks in cardiac development [14].
Dmef2
Dmef2 is expressed in all muscle-myosin expressing cells including the cardiac cells after the mesodermal subdivisions. Dmef2 is likely a direct target of tinman [14].
Heartless
The heart less gene of Drosophila melanogaster encodes an FGF receptor that is necessary for cardiac development. Heartless is expressed along with tinman in all mesoderm and allows early mesodermal cells to migrate dorsally to come in contact with decapentaplegic expressing ectoderm, which is a prerequisite for cardiac induction [14].
Slit
The EGF-repeat-containing protein, Slit is expressed in the cardiac cells of the heart. This is required for correct assembly of cardiac cells in the tube and has no role in the cardiac differentiation [14].
Hand gene
It encodes a highly conserved b HLH transcription factor expressed in heart cells. Hand genes were found in a variety of vertebrate species including humans [14,15].
The GATA gene pannier
It is required for cardiac cell formation while repressing the overproduction of a pericardial cell type. Pannier gene functions as a cardiac identity gene because its forced expression results in supernumerary cardiac cells. Pannier works synergistically with the Tinman in the activation of a heart enhancer for the Dmef 2 differentiation gene and the specification of the cardio blast fate [14].
In vertebrates, an understanding of the molecular basis of cardiac differentiation was hampered due to lack of suitable in vitro systems. The important role of tinman in Drosophila melanogaster prompted researchers to identify homologs of tinman in vertebrates. It is the earliest known marker for the cardiac lineage in vertebrates. Research is ongoing to establish a link between the fly genetic network and the vertebrate network. It has been shown that tin null mutants fail to specify the cardiac cells. Similarly the ortholog of tin in vertebrate is nkx 2.5 which is also associated with various cardiac defects. Thus understanding of genes and their function is critical in defining mechanisms of cardiac development and disease in higher organisms.
Thus all these findings suggest that Drosophila melanogaster may be a good source for identifying and isolating genes involved in cardio genesis and form the basis of cardio genic evolution. In this project, an attempt has been made to identify all the motifs in the enhancer sequences and filtering out the motifs which are potential transcription factor binding site. Once the motifs are identified they are mapped on the enhancer sequences. Further, using these mapped enhancers we can find other putative enhancer sequences, to determine unknown Drosophila melanogaster cardiac regulatory mechanism.
Materials and Methods
Identification of cardiac enhancer sequences
The enhancer sequences for the genes involved in cardiac development and differentiation in Drosophila melanogaster were retrieved from Red fly database Regulatory Element Database for Drosophila melanogaster v3.2 [16].
Motif Discovery using MEME
The motifs for these enhancer sequences were discovered using MEME (Multiple Em for Motif Elicitation). Input given was a text file containing the sequences of enhancer and output received the motifs within individual enhancers. MEME represents motifs as position-dependent letter-probability matrices which describe the probability of each possible letter at each position in the pattern [17].
Motif comparison with the known motifs using TOMTOM
After the discovery of all the motifs in respective enhancer sequences found in MEME, TOMTOM (a motif comparison tool) [19] was run on all the motifs to identify those motifs which are involved in transcriptional regulation. Tomtom is a tool for comparing a DNA motif to a database of known motifs. Tomtom contains all the motifs known in an organism for which a defined Gene Ontology has been described. This comparison between motifs is essential to find out all the functional motifs in our enhancer sequence and discarding the motifs without function.
Mapping of putative TFBS on dataset using MAST
The Transcription factors binding sites motifs identified by motif matching and are functionally relevant in organism's mechanism, were then mapped on our dataset of enhancer sequences using MAST [21]. MAST is a tool for searching biological sequence databases for sequences that contain one or more of a group of known motifs. MAST takes as input a file containing the descriptions of one or more motifs and searches a sequence database that you select for sequences that match the motifs. Once, the relevant motifs are mapped, our dataset now contains the training set for classifier to be used in machine learning process. The classifier will use the mapped TFBS on enhancers to find other sequences in the genome of Drosophila melanogaster that contains exactly the same set of TFBS motifs.
Results and Discussion
Sequence retrieval
A total of 17 cis regulatory module sequences were retrieved using REDFLY database. The RED fly is the most comprehensive available resource for experimentally validated cis-regulatory modules and transcription factor binding sites among the metazoan, along with their DNA sequence, their associated genes, and the expression patterns they direct. The sequences that have been tested by reporter gene assays in transgenic animals, and on binding sites discovered by DNase I foot printing and electro phoretic mobility shift (gel shift) assays are considered [16].
The sequences were retrieved based on their expression in the particular developmental stage and an excel file was created indicating the CRMs, their genes, their coordinates and the length of the sequences (Table1).
![]()
Bacterial species
Number of bacterial colonies on plate (cfu) x103 (control)
Number of bacterial colonies (cfu) x 103 grown on plate in the presence of silver nanoparticles (AgNPs)
AE [%] (percentage antibacterial activity)
100 �g cm-3 of AgNPs
B.subtilis subsp. spizizenii
252
187
25.80
S. aureus
510
382
25.10
P. aeruginosa
784
518
33.93
Table 1: Effect of silver nanoparticles synthesized by cell free extract of Myxococcus virescens on bacterial growth on agar medium.
These sequences were then used as input in MEME suite for motif discovery. A motif is a three-dimensional structural unit formed by a particular sequence of amino acids, found in proteins and which is often linked with a particular function [18]. Various parameters were set which were the maximum and minimum motif length, the number of motifs to identify and the strand on which it is to be identified. The MEME Suite web server provides a unified portal for online discovery and analysis of sequence motifs representing features such as DNA binding sites and protein interaction domains. Training set containing a group of protein sequences were taken as input to give motifs as output in MEME. The MEME results consist of: A description of the sequences in the "training set" showing the name, "weight" and length of each sequence, the occurrences of the motif sorted by p-value and aligned with each other. MEME also provides an opportunity to visualize motifs as block diagrams of the occurrences of the motif within each sequence in the training set [17] (Figure 2).
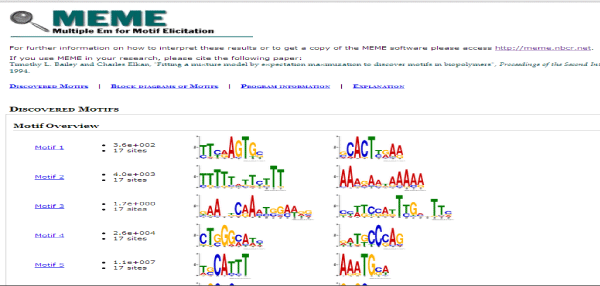
Figure 2: Result page of MEME, showing all the identified motifs in our dataset. Amino acid position specific scores are given in the form of logos and PSSM.
The motifs identified from MEME comprise of all the sequences sites regardless of their functional relevance. Many of these motifs do not have a Gene Ontology id. The Gene Ontology project provides ontology of defined terms representing gene product properties. The ontology covers three domains: cellular component, the parts of a cell or its extracellular environment; molecular function, the elemental activities of a gene product at the molecular level, such as binding or catalysis; and biological process, operations or sets of molecular events with a defined beginning and end, pertinent to the functioning of integrated living units: cells, tissues, organs, and organisms [19]. These motifs when compared to a database of known motifs categorized by the transcription factor families, results in the filtering of the query motifs as putative TFBS motifs.
A JASPAR CORE insect was used as a database of known TFBS motifs. It is an open source database comprising 126 motifs. JASPAR is the largest open-access database of matrix-based nucleotide profiles describing the binding preference of transcription factors from multiple species. The JASPAR database holds collections of PFM nucleotide profiles based on published experiments from diverse sources. The most widely used JASPAR collection is JASPAR CORE, which is a curated non-redundant set of TFBS profiles for multi cellular eukaryotes, based on experimental evidence. The JASPAR database aims to provide DNA binding profile per TF, as assessed by expert curators [20].
The TFBS motifs in query motif database were identified by aligning the target and query motif. Best hit was compiled based on the e value and p value by using TOMTOM (motif comparison tool). TOMTOM quantifies the similarity between two motifs, provides a numeric score for the match between two motifs and an estimate of the statistical significance of the score [18] (Figure 3).
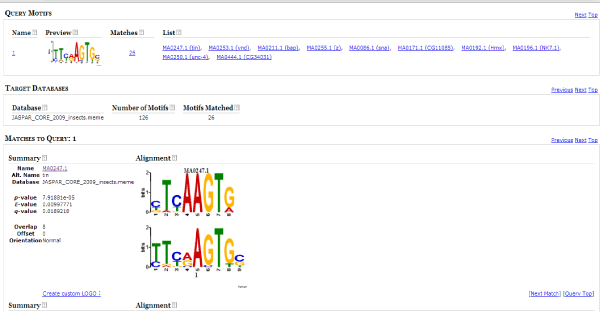
Figure 3: Result page of TOMTOM. Each motif from MEME was aligned against known motifs from JASPAR CORE insects.
Only matches for which the significance is less than or equal to the threshold set by the -thresh switch will be shown. By default, significance is measured by q-value of the match. The q-value is the estimated false discovery rate if the occurrence is accepted as significant. Only the alignments with the lowest e value score were considered and query motif corresponding to that score were then documented in a table showing the alignment along with the TFBS identifier.
Figure 4 lists out all the putative transcription factor binding motifs present in query motif database. These putative motifs were then mapped onto the enhancer sequence dataset using MAST (Motif Alignment and Search Tool) [21]. MAST works by determining the best match in the sequence to each motif. The scores for these best sequence motif matches are combined into a score for the overall match between the complete motif set and the sequence, resulting in an E-value for each sequence. The output from MAST is a list of the sequences for which the E-value is less than a user-specified threshold. The output also contains a block diagram showing the relative positions of the best motif matches in the high scoring sequences, and annotated alignments of the best motif matches. The match score of a motif to a position in a sequence is the sum of the score from each column of the position-dependent scoring matrix corresponding to the letter at that position in the sequence.

Figure 4: Table showing all the putative TFBS motifs along with their alignment from known database.
MAST results are compiled in Figures 5-13 showing the mapping of motifs. Illustrations in the figures show which motif is present and what is the e value of the prediction. In the present study, we found out the putative transcription factor binding site motif conserved in the sequences, these motifs were further mapped onto enhancer sequences which are a very important step towards discovering various unknown TF and TFBS involved in regulating transcription.

Figure 5: Various motifs were given different colours so that when mapped their identification becomes easier.
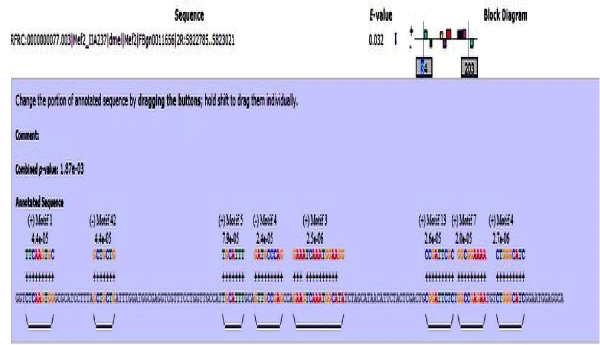
Figure 6: Mef2 enhancer sequence with the TFBS motifs.
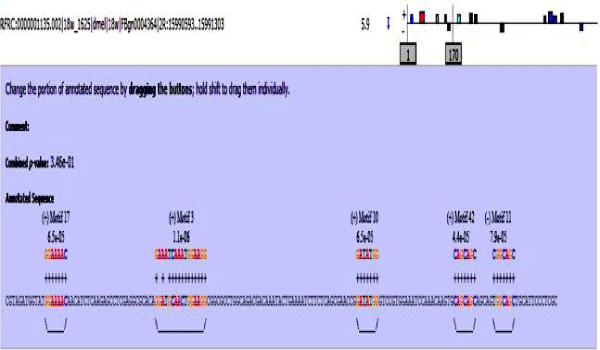
Figure 7: 18 wheeler sequence with mapped motifs.
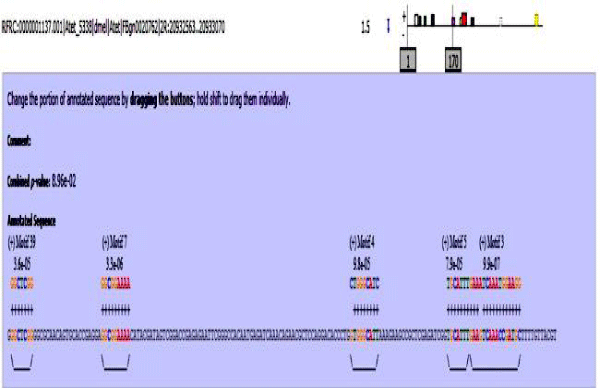
Figure 8: Atet enhancer sequence with mapped motifs.
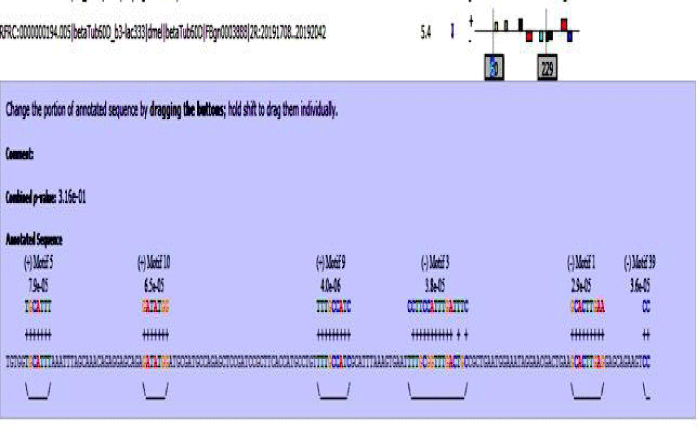
Figure 9: Beta tubulin 60D enhancer sequence with mapped motifs.
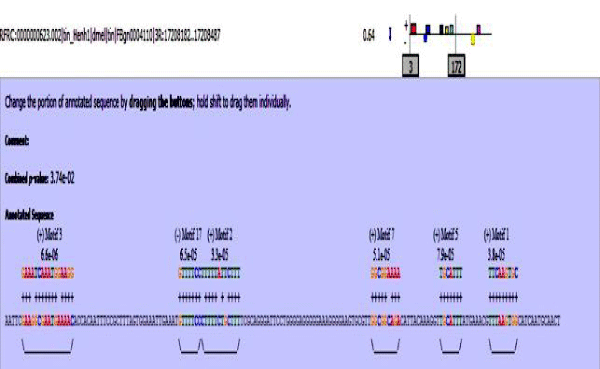
Figure 10: Tin enhancer sequence with mapped motifs.
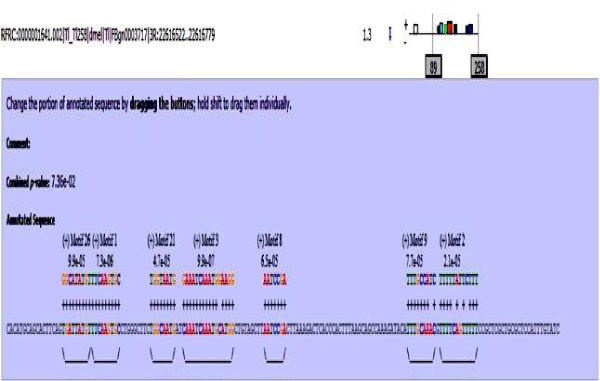
Figure 11: Toll enhancer sequence with mapped motifs.
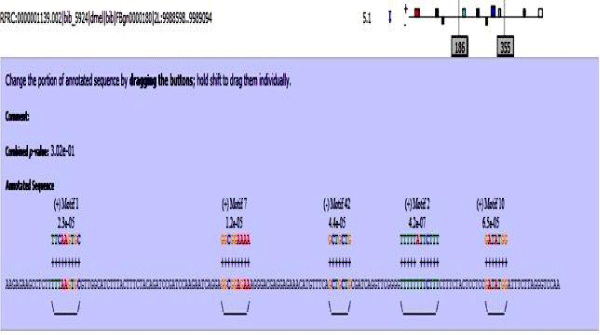
Figure 12: Bib enhancer sequence with mapped motifs.
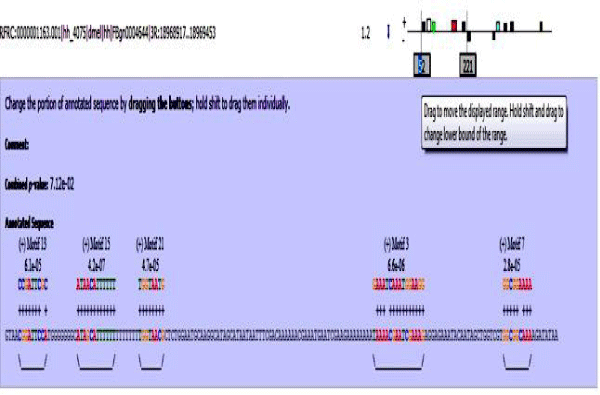
Figure 13: hh enhancer sequence with mapped motifs.
Conclusion
Heart is the first organ to form during embryogenesis and it is a vital organ in maintaining an organism haemostasis by pumping blood to every organ. Abnormities in the development and functioning of heart is a cause of many congenital heart disease, thus it is very important to characterize various genes and the regulatory network involved in heart functioning.
Previous studies have validated those genes which are evolutionary conserved show conservation in their functions also. Thus, the analysis of development in model organisms has provided important insight into developmental mechanisms [22,23]. Regulatory genes are known to function as components of regulatory networks; and there is increasing evidence that the genetic networks have also been conserved through evolution. Many of these genes also appear to have similar functions in Drosophila and in vertebrates. As described previously, drosophila gene tinman and its homologue in vertebrate nkx2.5, both are essential in development and functioning of cardiac cells [24, 25].
The spatial and temporal manifestation of these processes implies a complex program of genetic control. This genetic regulation is through precisely controlled processes of cell-cell signalling and regulators of gene expression. These processes were discovered through embryological studies. Recently, genetic screens in the zebra fish system have provided a link toward the identification of regulatory components in cardio genesis. Further, genetic and molecular studies of mesodermal tissue development the fruit fly Drosophila have also been instrumental in the identification of specific genes and processes in cardio genesis that appear to be conserved in all higher animals. Apart from tinman homologues, other genes can also be evolutionary conserved and present in higher organisms. Research towards this area is ongoing [26].
In the present work, we identified putative transcription factor binding motifs on the enhancer sequences of genes participating in the development and differentiation of Drosophila heart. These mapped sequences are of great importance in finding out other regions within the genome having the similar or identical set of motifs. The importance of having these motifs mapped is the knowledge about which transcription factor will bind to particular sequence. The interconnection between the sequences and transcription factor regulate the gene expression. By further extending this work to find other regions in whole genome, unknown mechanism involving TF-gene regulation can be unmasked, and will lead to increased knowledge about the genetic regulation within an organism and help in understanding the molecular and developmental functions of signalling processes during early cardio genesis that have been defined in both vertebrate and invertebrate models.
References
- Busser BW, Taher L, Kim Y, Tansey T, Bloom MJ, Ovcharenko I, et al. A machine learning approach for identifying novel cell type-specific transcriptional regulators of myogenesis. See comment in PubMed Commons below PLoS Genet. 2012; 8: e1002531.
- Ong CT, Corces VG. Enhancers: emerging roles in cell fate specification. See comment in PubMed Commons below EMBO Rep. 2012; 13: 423-430.
- Girardot C. Deciphering enhancer activity in Drosophila based on transcription factor occupancy and chromatin state chromatin state characterization (Doctoral dissertation, Université Pierre et Marie Curie-Paris VI). 2012.
- Bryantsev AL, Cripps RM. Cardiac gene regulatory networks in Drosophila. See comment in PubMed Commons below Biochim Biophys Acta. 2009; 1789: 343-353.
- Berman BP, Pfeiffer BD, Laverty TR, Salzberg SL, Rubin GM, Eisen MB, et al. Computational identification of developmental enhancers: conservation and function of transcription factor binding-site clusters in Drosophila melanogaster and Drosophila pseudoobscura. Genome Biol. 2004; 5: R61.
- Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. See comment in PubMed Commons below Proc Natl Acad Sci U S A. 2002; 99: 757-762.
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, et al. Genome-wide discovery of human heart enhancers. See comment in PubMed Commons below Genome Res. 2010; 20: 381-392.
- Bodmer R, Venkatesh TV. Heart development in Drosophila and vertebrates: conservation of molecular mechanisms. See comment in PubMed Commons below Dev Genet. 1998; 22: 181-186.
- Bodmer R. Heart development in Drosophila and its relationship to vertebrates. See comment in PubMed Commons below Trends Cardiovasc Med. 1995; 5: 21-28.
- Aerts S, van Helden J, Sand O, Hassan BA. Fine-tuning enhancer models to predict transcriptional targets across multiple genomes. See comment in PubMed Commons below PLoS One. 2007; 2: e1115.
- Gajewski K, Choi CY, Kim Y, Schulz RA. Genetically distinct cardial cells within the Drosophila heart. See comment in PubMed Commons below Genesis. 2000; 28: 36-43.
- Busser BW, Gisselbrecht SS, Shokri L, Tansey TR, Gamble CE, Bulyk ML, et al. Contribution of distinct homeodomain DNA binding specificities to Drosophila embryonic mesodermal cell-specific gene expression programs. See comment in PubMed Commons below PLoS One. 2013; 8: e69385.
- Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcriptional network. See comment in PubMed Commons below Dev Biol. 2002; 246: 14-28.
- Sellin J, Albrecht S, Kölsch V, Paululat A. Dynamics of heart differentiation, visualized utilizing heart enhancer elements of the Drosophila melanogaster bHLH transcription factor Hand. See comment in PubMed Commons below Gene Expr Patterns. 2006; 6: 360-375.
- Fossett N, Zhang Q, Gajewski K, Choi CY, Kim Y, Schulz RA. The multitype zinc-finger protein U-shaped functions in heart cell specification in the Drosophila embryo. See comment in PubMed Commons below Proc Natl Acad Sci U S A. 2000; 97: 7348-7353.
- Gallo SM, Gerrard DT, Miner D, Simich M, Des Soye B, Bergman CM, et al. REDfly v3.0: toward a comprehensive database of transcriptional regulatory elements in Drosophila. See comment in PubMed Commons below Nucleic Acids Res. 2011; 39: D118-123.
- Timothy L. Bailey, Charles Elkan. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. 1994; 2: 28-36.
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. See comment in PubMed Commons below Genome Biol. 2007; 8: R24.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. See comment in PubMed Commons below Nat Genet. 2000; 25: 25-29.
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. See comment in PubMed Commons below Nucleic Acids Res. 2014; 42: D142-147.
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. See comment in PubMed Commons below Bioinformatics. 1998; 14: 48-54.
- Kazemian M, Zhu Q, Halfon MS, Sinha S. Improved accuracy of supervised CRM discovery with interpolated Markov models and cross-species comparison. See comment in PubMed Commons below Nucleic Acids Res. 2011; 39: 9463-9472.
- Ivan A, Halfon MS, Sinha S. Computational discovery of cis-regulatory modules in Drosophila without prior knowledge of motifs. See comment in PubMed Commons below Genome Biol. 2008; 9: R22.
- Lien CL, McAnally J, Richardson JA, Olson EN. Cardiac-specific activity of an Nkx2-5 enhancer requires an evolutionarily conserved Smad binding site. See comment in PubMed Commons below Dev Biol. 2002; 244: 257-266.
- Philippakis AA, Busser BW, Gisselbrecht SS, He FS, Estrada B, Michelson AM, et al. Expression-guided in silico evaluation of candidate cis regulatory codes for Drosophila muscle founder cells. See comment in PubMed Commons below PLoS Comput Biol. 2006; 2: e53.
- Khan MA, Soto-Jimenez LM, Howe T, Streit A, Sosinsky A, Stern CD. Computational tools and resources for prediction and analysis of gene regulatory regions in the chick genome. See comment in PubMed Commons below Genesis. 2013; 51: 311-324.