
Research Article
Austin J Biotechnol Bioeng. 2014;1(4): 9.
Cloning and Expression of Biologically Active Buffalo Thyroid Stimulating Hormone (Butsh) in Methylotropic Yeast Pichiapastoris
Nidhi Vashistha1, Rajan Dighe2, Taruna Arora1, Nikki Kumari1 and Kambadur Muralidhar3*
1Department of Zoology, University of Delhi, India
2Department of Molecular reproduction, Indian Institute of Science, India
3Department of Life Sciences & Biotechnology, South Asian University, India
*Corresponding author: Kambadur Muralidhar, Department of Life Sciences & Biotechnology, South Asian University, #230 Akbar Bhawan, Chanakyapuri, New Delhi-110021, India.
Received: August 25, 2014; Accepted: September 26, 2014; Published: September 26, 2014
Abstract
Superovulation and Embryo transfer are commonly used techniques for improving genetic potential and disseminating superior germ plasm. The overall results of superovulation and embryo transfer have been discouraging in buffalo. Unfortunately, animals are notably unresponsive to exogenously administered hormones as only heterologous preparations have been used since homologous preparations are not available. Thus, it was of primary importance to develop effective homologous preparations which could be used in field trials. An in vitro assay was standardized to assess the TSH bioactivity. The effect of stimulation of buffalo thyroid follicles by bTSH was measured in the form of released cAMP. Total RNA was extracted from the pituitary and first strand cDNA was made. The cDNA for the specific subunits were amplified using gene specific primers. Heterodimeric buTSH was cloned and expressed in yeast Pichia pastoris. Successful co-expression of the TSH subunits was achieved with an average yield of 5-7 mg/L of the heterodimeric hormone. The recombinant hormone was secreted into the fermentation medium since the hormone subunits were cloned downstream to the yeast Α mating peptide. The recombinant hormone was further purified by hydrophobic interaction chromatography using Phenyl- Sepharose and characterized using similar homologous and heterologous assay systems. The recombinant hormone was active in ex-vivo bioassay and the bioactivity of the purified recombinant buTSH was found to be 8 IU/mg of protein.
Keywords: Buffalo; Thyrotropin; Recombinant TSH; Pichia expression
Introduction
Owing to their potential therapeutic applications, the production of recombinant glycoprotein hormones of pituitary origin from farm animals like sheep, cattle, buffaloes etc. in large quantities, is an important research goal [1,2]. The Thyrotropin family of hormones comprise Follicle Stimulating Hormone (FSH), Luteinizing Hormone (LH) and Thyroid Stimulating Hormone (TSH). Each of them comprises an Α and a β subunit, where Α is species specific and β is hormone specific [3]. The individual subunits are biologically inactive while hetero dimeric protein is active. Two major factors which influence the process of correct folding of the hormonal subunits are disulfide bridge formation and glycosylation. These considerations govern the choice of expression system for the hyper-expression of glycoprotein hormones.
Expression of the recombinant glycoprotein hormones had been attempted using mammalian and insect cell expression systems [4- 7]. However, due to the requirements of complex media, as well as, sophisticated cell culture facilities, scaling-up of expression was not very cost effective. The requirement of glycosylation for correct folding of the hormone subunits limits the use of bacterial expression system where only a non-glycosylated protein is obtained. The yeast expression system using methylotropic yeast, Pichia pastoris, was found to be an ideal system for hyper-expression of glycoprotein hormones as it blends the advantages of both prokaryotic as well as, eukaryotic expression systems. Using this expression system, the members of glycoprotein hormone family and their derivatives have been expressed by a number of groups [8-12]. The strategy is simple. Alcohol oxidase, encoded by AOX gene is the first enzyme in the ethanol utilization pathway, accounting for 35% of total cell protein in Pichia cells grown on a limited amount of methanol. So the gene of interest is cloned under the regulation of an inducible AOX1 promoter resulting in controlled expression. As the foreign gene is randomly incorporated into the host genome the clones are genetically stable and growth can be scaled up without loss of yield by manipulating the gene copy number integration. The alpha mating factor signal sequence is fused with the gene of interest and is used for the efficient secretion of the protein into the media.Post-translational modifications are very similar to the mammalian system but not identical.
The yield of native buffalo TSH (buTSH) when purified biochemically from slaughter house material is reportedly very low [13]. Hence heterodimeric buTSH was cloned, expressed and purified. The recombinant buTSH was biologically active.
Materials and Methods
BovineTSH (bTSH) reference preparation and Ovine TSH (oTSH) antiserum were obtained from NHPP through Dr. A. F. Parlow. BuTSH used as a reference and control was purified in the laboratory through biochemical methods [14]. The bacterial strain used for the propagation of the plasmid, were DH5Α and were procured from Gibco BRL, USA. The plasmid used for DNA manipulations, pBlue script SK+ was obtained from Stratagene (LA Jolla, CA, USA). cAMP and mono succinyl adenosine 3', 5' - monophosphoric acid tyrosine methyl ester (ScAMPTME) were obtained from Sigma Ultrapure Chemicals, USA. The tissue culture-ware was purchased from Nalgene, USA and the tissue culture media were purchased from Gibco-BRL, USA.All the restriction enzymes and modifying enzymes required were purchased from Boehringer Mannheim, GHBH, Germany; or from MBI Fermentas. Yeast extracts, peptone, yeast nitrogen base without amino acids (YNB) required for growing Pichia pastoris cells were obtained from Invitrogen Corp, CA, USA. Phenyl Sepharose, Concanavalin A Sepharose, Cibacron Blue and Reactive Yellow Sepharose matrices used for purification and characterization of the recombinant proteins were purchased from Sigma Chemical Company, St Louis, MO, USA.All other reagents were obtained from Sigma Chemical Company, St Louis, MO, USA.
General methodologies
All the DNA manipulations were according to standard techniques [15]. The bacterial vectors PBSK+ and PICZa were grown in LB medium. Plasmids were isolated from the positive clones either by the miniprep or midiprep by standard protocols. The isolated plasmid DNAs were run on gel to check their quality and were purified after restriction enzyme digestion as per standard protocols. The cDNA encoding the mature secreted form of buTSH Α and β (without their cognate signal peptide) was PCR-amplified using the buffalo pituitary cDNA (made by Reverse Transcription of total RNA) as the template and the primers that were designed to introduce Xho I and Not I sites at the 5' and 3' ends of cDNA respectively.
The forward primer for Α subunit:
5' CC CTC GAG AAA AGA TTT CCT GAT GGA GAG TTT ACA ATG CAG GGC 3'
Reverse primer
5' TT GCGGCCGC TTA GGA TTT GTG ATA ATA ACA AGT ACT GC 3'
The forward primer for the β subunit:
5'CC CTC GAG AAA AGA TTT TGT ATT CCA ACT GAG TAT ATG ATG CAT GTC G 3'
Reverse primer
5' TT GCGGCCGC TTA GAT AGA AAA TCC CAC CAT ATA GGA CTT CTG A 3'
The PCR amplified product was cloned into pPICZa expression vector at the Xho I and Not I sites to obtain pPICZabuTSHΑ and pPICZabuTSHβ after intermediate cloning into the bacterial vector, pBlue script and confirming the sequence by automated DNA sequencing. Further, the positive clones were checked by PCR and restriction enzyme digestion before the yeast cells were transformed.
Preparation of the recombinant plasmid
One of the positive clones of each pPICZabuTSH Α and pPICZabuTSH β were grown in 100 mL LB broth and the plasmid was isolated. The final plasmid pellet was resuspended in TE buffer. This recombinant plasmid was then linearized using Pme I enzyme. The final linearized plasmid was again re-precipitated and stored in sterile water till further use.
Preparation of X-33 competent cells
Pichia pastoris (X33) was grown to A600 of 0.8-1.0 in 50 mL of 1% yeast extract, 2% peptone and 2% dextrose (YPD) at 30°C in a shaking incubator. The cells were harvested by centrifugation at 500g for 5 min at room temperature and resuspended in 9 mL of ice cold BEDS [10mM Bicine, NaOH, pH 8.3, 3% (v/v) ethylene glycol, 5% (v/v) Dimethyl sulfoxide (DMSO), and 1 M sorbitol] solution supplemented with 1 mL of 1 M Dithiothreitol (DTT), incubated for 5 min at 100 rpm at 30°C in a shaking incubator. Further, the cells were washed and resuspended in 1 mL (0.02 volumes) of BEDS solution without DTT. Aliquots (40 uL) of the competent cells were immediately made and kept at -70°C. Preferably competent cells were made fresh and not stored beyond a week.
Transformation by electroporation
An aliquot of the competent cells was transferred to a 2 mm gap electroporation cuvette (Molecular Bio Products) for transformation and mixed with 4 uL (containing 50-100 ng) of the Pme I digested expression cassette obtained from the recombinant plasmid and incubated on ice for 15 min following which the cells were subjected to electroporation in a Gene PulserR II electroporator (Bio-Rad Laboratories, Hercules, CA, USA) using a charging voltage of 1500 V; resistance, 200 Q; capacitance, 25 pF for about 4-6 msec. Immediately after electroporation, 1 mL of 1 M sorbitol was added to the cuvette and the contents were transferred to a separate tube and incubated at 30°C without shaking for 1 h for recovery. Several volumes (25, 50, 100, 200 uL) of the cell suspension were placed on selected YPD plates containing 100 ug/mL Zeocin and incubated at 30°C for 2-3 days till colonies appeared.
Screening of the transformants
The Pichia transformants obtained as described above were screened for their ability to secrete heterodimeric buTSH upon induction with methanol as the carbon source. The individual clones were first grown in 5 mL YPD broth for 48 h at 30°C. The cells were centrifuged and resuspended in 1.34% YNB (without amino acids), 100 mM Potassium phosphate buffer, 0.5-1% methanol (YPM) and were further grown at 30°C for another 48-72 h with addition of 1% methanol every 12 h. The supernatants of the induced cultures were then assayed for protein expression using specific radio- receptor assay as described earlier. The clone secreting maximum levels of heterodimeric buTSH was used to scale up.
Radio-iodination by iodogen method
The radio iodination of bTSH was carried out using the Iodogen method and was characterized according to Dighe et al. [16]. The specific activity of the probes was calculated using TCA precipitation method and was in the range of 100,000 cpm/ng of protein.
The hTSH Receptor cells grown in a 21 cm culture dish were washed with chilled 0.05 M Tris-HCl, pH 7.4 containing 5 mM MgCl2 (RRA Buffer). The cells were scraped from the plate in 5 mL of RRA buffer containing protease inhibitors (5 mM PMSF, 0.02ug/ mL Aprotinin) and homogenized with 10 strokes in a Dounce homogenizer with a loose fitting pestle. To the homogenate, equal volume of 0.6 M sucrose was added and the lysate was centrifuged at 10,000g for 30 min. The pellet containing the total membrane preparation was washed once with 1X RRA buffer without sucrose, aliquoted and stored in RRA buffer containing 20% glycerol and protease inhibitors in -70°C.
Radio-receptor assay
Approximately 5 ug of total membrane preparation was incubated with different concentrations of the yeast culture supernatant from different colonies for 2 h at room temperature in a total reaction volume of 250 uL. The bound hormone was separated from the free by precipitation of the hormone-receptor complex with 2.5% PEG at 4°C and centrifugation at 5000g at 4°C for 20 min. The supernatant was discarded and the bound radioactivity was determined in a Perkin Elmer auto gamma counter. The non-specific binding was determined by incubating the membrane preparation with the labeled probe in the presence of excess of cold TSH (400 ng/mL).
Shake flask culture
0.5 mL of 48 h grown culture was inoculated into 50 mL YPD medium. This was grown to saturation and added to 2 L YPD broth (2% inoculum) and was incubated at 30°C for 48 h. The cells were then pelleted and resuspended in half volume i.e., 1L of BMMY (induction medium) and were grown for further 72 h. The supernatant was collected and was used for buTSH purification and characterization using Con A, Blue Sepharose and Reactive Yellow matrices. The recombinant buTSH was finally purified using Phenyl Sepharose and closely monitored using ELISA.
Purification
Con A Sepharose chromatography: Pre-swollen Concanavalin A-Sepharose gel was packed into a 20 mL glass column. The column was equilibrated with 0.01 M Tris-HCl buffer (pH 7.5), 0.3 M sodiumchloride and 0.001 M each of CaCl2, MnCl2, and MgCl2 (Buffer 1). Supernatant of an induced yeast culture was loaded onto the column. After the sample descended into the column the flow was stopped for 30 min to allow protein interaction with the matrix. Then the column was washed thoroughly with the loading buffer and the bound proteins were sequentially elutedusing 0.01 M Tris-HCl buffer (pH 7.5), 0.3 M sodium chloride (Buffer 2), 0.01 M Tris-HCl buffer (pH 7.5) 0.3 M sodium chloride, 0.05 M a-methyl-D-Mannoside (Buffer 3), 0.01 M Tris-HCl buffer (pH 7.5) 0.3 M sodium chloride, 0.3 M a-methyl-D-Mannoside ( Buffer 4) and then with 0.05 MTris-acetate buffer (pH 5.0) and 1 M sodium chloride (Buffer 5). Fractions (2.0 mL) were collected with a flow rate of 15 mL/h. The absorbance at 280 nm and immune reactivity of each tube was determined (direct binding ELISA).
Reactive yellow chromatography: Swollen agarose beads cross linked to Reactive Yellow 86 dye (Sigma) were packed in a 1 mL column. The column was equilibrated with Citrate Buffer (25 mM, pH 5.0) at the rate of 15 mL/h. The induced yeast medium having the recombinant buTSH was loaded on to the column. Fractions (1 mL) of unbound and bound protein were collected in the test tubes. The subsequent elution was done using Phosphate buffer (50 mM, pH 6.0), phosphate buffer (50 mM, pH 7.0), Phosphate buffer (50 mM, pH7.0) with 1M NaCl and with 80% ethylene glycol. All the collected fractions were assessed for their absorbance (280 nm) and immune reactivity. A graph was plotted between fraction numbers and their respective absorbance and immune reactivity.
Cibacron blue chromatography: A 1 mL column of Cibacron Blue 3GA agarose gel was prepared, equilibrated with 810 bed volumes of 50 mM phosphate buffer, pH 7.0. The column was maintained at a constant flow rate of 20-25 mL/h. The sample dissolved in the equilibration buffer was loaded onto the gel. After the sample entered the gel the flow was stopped. Thirty min later the gel was washed with the same buffer at a constant flow rate to wash all the unbound proteins. Subsequently the bound proteins with ionic interactions were eluted with 50 mM PB pH 7.0 containing 1 M NaCl. Finally the column was washed with 80% ethylene glycol, to elute out the hydrophobic protein fraction bound to the column. Fractions (1 mL each) of unbound and bound protein were collected and their absorbance was measured at 280 nm and the immuno reactivity was also checked against anti oTSH a/s.
Hydrophobic interaction chromatography: The Pichia culture supernatant was mixed with solid ammonium sulfate to achieve the final concentration of 1.6 M and was buffered with 50 mM Phosphate buffer, pH 7.4. Any precipitate formed was removed by centrifugation at 22,000g and the supernatantwas applied to Phenyl Sepharose column (bed volume 20 mL (15 mL/min flow rate) previously equilibrated with the same buffer (1.6 M ammonium sulfate, 50 mM phosphate buffer, pH 7.4). The column was washed with 5 column volumes of the equilibration buffer. The bound proteins were sequentially eluted with 50 mM phosphate buffer pH 7.4, 10%, 20% and 50% acetonitrile in 50 mM phosphate buffer. The absorbance at 280nm was recorded for all the fractions. Different fractions obtained were assayed for hormone specific immuno reactivity using TSH specific antibodies. All the samples from the Con A, Reactive yellow, Blue Sepharose and HIC column were checked for its immunoreactivity against oTSH P antiserum using direct binding ELISA.
Homologous bioassay (ex situ) using buffalo thyroid follicles
Buffalo thyroids were homogenized in KRBG (10% homogenate) containing theophylline. The homogenate (100 uL) was incubated for 30 min at 37°C with varying concentrations of the standard bTSH and the recombinant buTSH clones' culture supernatant separately. The incubation was stopped by immersion of tubes in aboiling water bath followed by addition of 0.1 N HCl. Total cAMP was measured by competitive ELISA.
Heterologous bioassay using HEK-293TSH-R cells
The cells were plated in a 48 well plate and grown in DMEM supplemented with 10% FBS for 48 h (80-85% confluence) in a CO2 incubator at 37°C. The cells were washed once with DMEM and incubated in 100 uL of fresh medium containing 0.5 mM of general phosphodiesterase inhibitor Isobutyl Methyl Xanthine (IBMX) for 30 min at 37°C before the ligand(s) treatment. To this varying concentrations of standard bTSH and recombinant buTSH yeast culture supernatants was added and the cells were incubated for 15 min at 37°C. The incubation was stopped by lysing the cells by addition of 0.1 N HCl (200 uL) and total cAMP was measured by competitive ELISA.
Gel electrophoresis and immunoblotting
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (15% SDS-PAGE) was carried out with the purified proteins, both under reducing, as well as, non-reducing conditions. The protein bands in the gel were visualized by silver staining or with Coomassie blue staining. The proteins electrophoresed on SDS-PAGE were transferred to Nitrocellulose membrane and were probed with antisera specific for TSH or its subunits. The protocols followed for gel electrophoresis and Western blot analysis were the same as described by Laemmli [17] and Towbin et al [18].
Staining for glycosylated proteins
To determine the glycosylation status of the Pichia expressed hormones, the proteins electrophoresed on SDS-PAGE were stained for their carbohydrate using rapid periodic acid Schiff (PAS) staining procedure. The protein samples in the gel were fixed by immersing the gel in 20% trichloroacetic acid for 10 min. Carbohydrates on proteins were oxidized by incubating the gel in 0.7% periodic acid in 5% acetic acid for 20 min. The gel was then washed with distilled water and was later transferred to the Schiff's reagent (neat) for 20 min. The reaction was stopped by transferring the gel in 5% solution of potassium bisulfite prepared in 5% acetic acid. Excess stain was removed from the gel by washing it with destaining solutions containing different grades of methanol and acetic acid, ranging from 5% methanol and 7.5% acetic acid to 50% methanol and 30% acetic acid till the gel regained its colorless transparent nature. The glycoproteins showed bright pink colored bands on staining.
Results
Cloning and expression of buffalo thyroid stimulating hormone
The cDNA encoding buTSHΑ and buTSHβ (without cognate signal peptide coding region) was PCR amplified from the buffalo pituitary cDNA using the gene specific primers and was found to be around 320 bp and 400 bp (results not shown). The nucleotide sequence was confirmed by automated DNA sequencing. Further, the buTSHΑ and buTSHβ coding sequence were cloned into Pichia expression vector pPICZaA (not shown), downstream of Α-mating factor signal peptide as it is observed that the expression of heterologous protein is much higher in presence of Α-mating factor signal peptide than with its cognate signal peptide [10].
The positive clones of pPICZaA for buTSH Α and β were again cross checked by PCR and restriction enzyme digestion. The plasmid was isolated in bulk amounts from the positive clones and was linearized using a selective enzyme from Pichia manual. There are a number of enzymes which can be used like Bgl II, Sac I, Pme I etc. (Easy select™ kit manual, Invitrogen). The criterion for using the enzyme is that the cutting site (restriction site) of that particular enzyme should not fall in the desired cDNA sequence which is to be cloned. There was no restriction site for Pme I in both the sequences; hence this was the chosen enzyme for linearization. The linearized plasmids were then electrophoresed on 1% agarose and were purified from the gel and resuspended in autoclaved MQ water.
Electroporation and screening of the transformants
The transformed yeast cells were plated on a Zeocin plate. The antibiotic resistance was slowly increased to find double transformants. The Zeocin concentration was increased sequentially from100 ug/mL to 1000 ug/mL. A few of the colonies were able to grow on the increasing concentration. However, some could not sustain the increased pressure of the antibiotic. The colonies which grew on highest concentration of Zeocin after 48 h were inoculated in YPD broth grown for 48 h and the protein of interest was induced using methanol in BMMY.
Radio-receptor assay
The level of expression of the recombinant heterodimeric TSH was determined by RRA. The radio-iodination of bTSH was performed essentially by Iodogen method. Initially 100 uL of each clone supernatant grown in BMMY and induced by methanol was added to HEK-293 hTSH-R membrane preparation which was made over a sucrose gradient. The displacement of the labeled bTSH was taken as a parameter of the buTSH secretion. The displacement of bTSH was plotted against the clone number (Figure 1). About 10 from the total 30 selected clones were found to be positive for these creation of a protein which was able to bind to the receptor. A few of the positive clones along with the negative controls were further used for a confirmatory test by incubating the membranes with different volumes of the supernatant (results not shown). This gave a dose response curve when increasing volumes of the yeast culture supernatant was used (Figure 2).
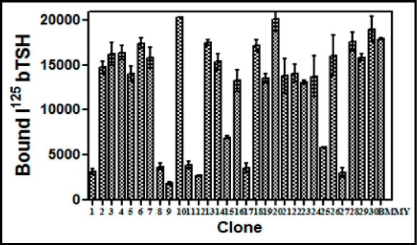
Figure 1: Screening and selection of buTSH expressing Pichia clones using radio-receptor assay. 100 pL of culture supernatants of different clones was incubated with 125I bTSH (200,000 cpm/tube) for 1 h at RT and followed by the addition of TSHR membrane preparation and the incubation was continued for another1 h at RT. The bound counts were plotted against the clone number. The data represent Means + SEM and the experiment was repeated thrice.
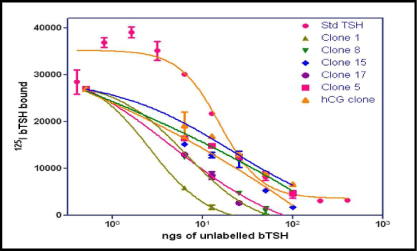
Figure 2: Results of radio receptor binding assay. Confirmation of the positive clones of buTSH. Increasing doses of bTSH or volume of the culture supernatant of the clones was incubated with the membrane receptor preparation along with radio labeled TSH and the bound radio activity measured. bTSH standard is also incubated under the same conditions. The bound counts are plotted as a function of increasing concentration.
Shake flask culture
The clone no #1 was used for the laboratory scale shake flask culture of the yeast to obtain recombinant buTSH. The clone was initially grown overnight in YPD broth and was further used as inoculum. The yeast was grown for 48 h and then was induced in BMMY for 72 h using methanol. Methanol was supplemented every 12 h later. The cells were then harvested by centrifugation and the supernatant was collected. The pH of the yeast supernatant was in a range of 5.9-6.7. The pH was then neutralized using liquid ammonia. Small volume of the media was dialyzed and lyophilized. It was used for total protein estimation and estimation of recombinant buTSH by ELISA (Figure 3).The total protein isolated from the pichia culture supernatant was observed to be in the range of 900 mg-1200 mg/L. The clone was secreting approximately 5-7 mg/L buTSH (shake culture level) and was selected for further purification and characterization.
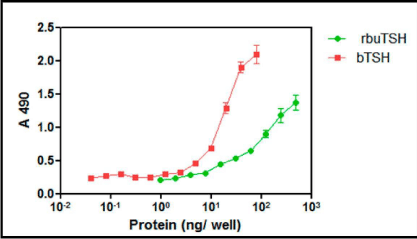
Figure 3: ELISA for Pichia expressed recombinant buTSH. The protein was coated on ELISA plate and TSH specific antibody was used to probe the hormone. The colour produced was measured at 490. The intensity of colour produced is plotted as a function of protein concentration.
Purification of recombinant buTSH
Con A Sepharose chromatography: The yeast expressed buTSH distributed itself in almost all the differentially eluted fractions as is evident from the Figure 4. Here about 18% was in unbound fraction (UB1 and UB2), 35% was weakly bound and 35% was tightly bound fraction. The results clearly indicated that the recombinant buTSH was differentially glycosylated as all the eluates carry the TSH immunoreactivity.
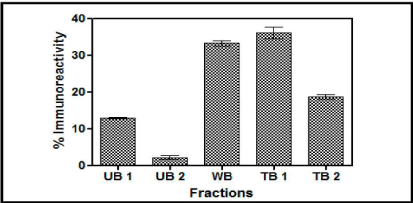
Figure 4: A. Chromatogram of the yeast supernatant when loaded on Con A column. The fractions were checked for the presence of immune reactive buTSH. The percentage immune reactive data depicts that the yeast expressed TSH in differentially eluted fractions. UB unbound, WB weakly bound and TB tightly bound.
Cibacron blue and reactive yellow chromatography: Similarly, the Cibacron Blue (Figure 5) and Reactive Yellow (results not shown) fractions were also screened as potential matrices for purification as they have been employed in the glycoprotein hormone purification earlier [19]. However, here it was again evident that these matrices were not able to purify the recombinant buTSH. The buTSH distributed itself in almost every differentially eluted fraction from Cibacron Blue (45% and 42%) and Reactive Yellow (42%, 22% and 25%). Hence none of the pseudo- affinity chromatography methods could be used for purifying the recombinant hormone.
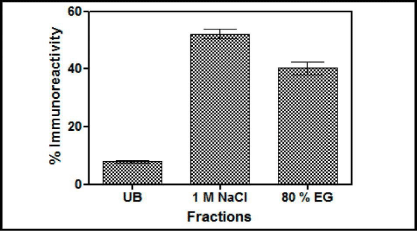
Figure 5: Results of chromatography of the yeast supernatant when loaded on Cibacron Blue- Sepharose column. The fractions were checked for the presence of immune reactive buTSH. Arrows indicate the buffer changes. The percentage immune reactive data indicates that the yeast expressedTSH gets distributed in both the salt elutable (1 M NaCl) and organic solvent elutable fractions. (80% Ethylene Glycol). UB shows the unbound fraction.
Hydrophobic interaction chromatography (HIC)
The conditions for maximum binding of the TSH to HIC matrix were already optimized in the lab by carrying out pilot experiments. Most of the TSH activity present in the yeast culture supernatant was adsorbed onto the matrix in presence of 1-1.6 M ammonium sulfate. The unbound proteins were washed with the loading buffer (50 mM PB containing 1.6 M ammonium sulfate). The maximum TSH activity after ELISA analysis was shown to be concentrated in the 50 mM PB eluate (Figure 6). The introduction of the organic solvent like acetonitrile was necessary to remove all the bound proteins from the column. The fractions having TSH activity were pooled and dialyzed. Hydrophobic interaction chromatography thus served the dual purpose of concentrating hormone activity and removal of salt from the culture supernatant in a single step.
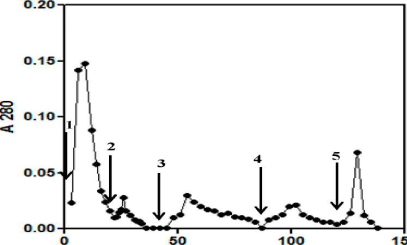
Figure 6: Chromatogram depicting the HIC purification of buTSH expressed in Pichia pastoris. The yeast culture medium was loaded on the Phenyl Sepharose column in presence of high salt. The bound proteins were differentially eluted. The absorbance at 280nm is plotted as a function of elution volume.
SDS-PAGE
The protein peak showing maximum TSH activity after concentration was subjected to an SDS-PAGE analysis to see the status of purified TSH. It was really difficult to separate the TSH straight from culture medium on the gel because the salt in the media interferes with the movement of proteins. However, the PB eluate of HIC column carrying maximum TSH activity clearly showed the two bands of Α and β subunits (results not shown). The gel was also stained to show the total carbohydrate staining. Hence the expressed recombinant buTSH is relatively free of any contaminating protein and is also glycosylated. The recombinant buTSH was also probed with an antibody generated towards whole TSH. The two subunits bands were seen very clearly.
Biological activity of recombinant buTSH
To demonstrate that recombinant buTSH is biologically active, its ability to bind to TSH receptor and elicit a biological response was next determined. As already shown by the RRA data that recombinant buTSH was able to bind to the TSH Receptor cell line. The ability to transduce the signal was demonstrated by homologous and heterologous assay wherein the purified recombinant buTSH from various clones was observed to cause cAMP release. End point was measured by estimating cAMP secreted into the medium. As shown in the Figure 7 and Figure 8, the recombinant buTSH could stimulate response both in vitro and in vivo. The bioactivity was found to be in the range of 8-9 IU/mg of the protein.
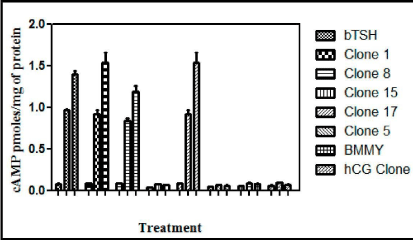
Figure 7: Thyroid follicle assay of the recombinant TSH. Standard bTSH or the yeast culture supernatants were added to a homogenate of the buffalo thyroid follicles. The release cAMPwas measure by competitive cAMP ELISA. The experiment was repeated thrice and ELISA was done in triplicates. The data is presented as Mean±SEM.
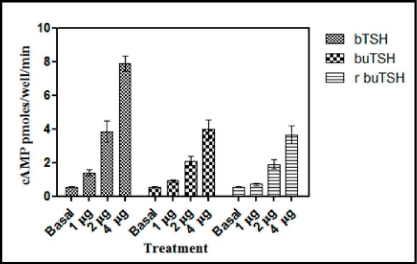
Figure 8: In vitro bioassay for the recombinant buTSH. The purified native and recombinant TSH were checked for their efficiency to release cAMP from the hTSHR cell lines. Different doses were added to HEK 293 cells expressing TSHR (pre-treated with 1mM IBMX) for 15 min. Cells were lysed with the addition of 0.2N HCl and cAMP produced was quantitated with competitive ELISA.
Discussion
The results presented here show that the recombinant buTSH produced by P. pastoris was biologically active as assessed by an in vitro bioassay. Moreover the recombinant hormone could bind to the receptor with a binding profile similar to that of native TSH indicating the recombinant hormone could also adopt a conformation resembling that of the native TSH.
Although not required for the binding to the receptor but N-linked glycosylation influences the bioactivity of glycoprotein hormones both in vivo and in vitro [20]. In the present study, recombinant hormone stimulated the cAMP production in homologous and heterologous assays, suggesting that the suitable glycosylation structures have been added to the recombinant protein during the Pichia secretory pathway [21]. PAGE analysis shows that the size of the heterodimeric buTSH was around 40 kDa. However the reducing PAGE showed that that the two subunits were around 22 and 25 kDa. The native page also shows a lower band which is likely to be a degradation product.
The glycoprotein hormones are interesting group of molecules, not only because of their importance in reproduction and overall physiology, but also because of their unique structural features. These glycoprotein hormones have proved to be interesting models for the study of protein folding, protein-protein interactions and role of carbohydrates in protein function. In addition, their potential therapeutic applications make them interesting and important from a commercial point of view.
The choice of expression system for expressing the glycoprotein hormones is guided by several important considerations. Glycoprotein hormone family has two important and unique structural features i.e., the two subunits Α and β are glycosylated in all the hormones and both of these subunits have multiple disulfide bonds. The biological action of the hormones is dictated and affected by the carbohydrate moiety; it has been studied in great detail. Deglycosylated (Chemically or enzymatically) heterodimeric hormones have been shown to be capable of binding to the receptor but incapable of further downstream signal transduction [22-26]. Also the glycosylation during biosynthesis is a precondition for correct folding of individual subunits [27-29]. The folding of β subunit is slower and the folding completes only after its assembly with Α subunit while folding of the Α subunit proceeds faster and co- translationally [30-32]. Also the slower folding of the β subunit has been thought to be the rate limiting step in the subunit assembly [30].
With reference to the second important structural feature (the presence of quite a few disulfide bridges in both subunits), three of these disulfide bridges in each subunit have been shown to be involved in the formation of a cystine knot structure. The importance of correct disulfide bond pairing, and that of the sequence of formation of disulfide bonds in hCG Α, is demonstrated by in vivo folding studies [33]. Hence, glycosylation of the subunits and disulfide bond formation are two simultaneous and totally interrelated processes. A vital role has been implicated to be played by the redox environment within the endoplasmic reticulum and the possible involvement of protein disulfide isomerase and cell- specific chaperones in the process of folding and association of glycoprotein hormone subunits [34,35]. These attributes render the folding of the glycoprotein hormone subunits a very important process. This process of correct folding is observed to be inefficient in vitro as in the case of recombinant protein expression systems. There are major problems associated with the expression of the hormonal subunits in the prokaryotic system [36]. Here the expressed protein is not glycosylated, and goes into inclusion bodies, requiring purification under very stringent denaturing conditions and subsequent refolding in vitro. Conversely, the mammalian expression system is capable of producing biologically acpossesses the ease of genetic manipulability and growth characteristics of prokaryotes and the subcellular machinery for post-translational modification of eukaryotes. The passage of the expressed protein through the secretory pathway allows post-translational modification events such as proteolytic cleavage and maturation, glycosylation and disulfide bond formation to occur. The methylotropic yeast, Pichia pastoris, has been used for high level expression of a wide variety of recombinant proteins [37- 39]. A critical factor for selection of this system has been the ease with which scaling-up from shake flask to high-density fermenter cultures is possible without loss of specific productivity. The existence of well-established fermentation procedures in a medium that is a defined mixture of salts, trace elements, biotin and carbon source, makes the system inexpensive and suitable for a high level of recombinant protein production. In Pichia pastoris, the presence of a tightly regulated methanol-inducible promoter allows the selective induction of the recombinant protein in the presence of methanol as the carbon source, thus providing the advantage of controlling growth and production phases separately and independently. Furthermore, integration of the expression cassette within the genome of the host maximizes the stability of foreign protein production strains. This strategy also allows the selection of strains that have multiple copies of expression cassette integrated into the genome and are hence capable of producing high levels of the recombinant protein [8]. In our studies, we also observed that the level of expression of buTSH subunits could be correlated to the number of expression cassettes integrated into the genome, and hence to the increasing resistance to Zeocin. The recombinant strains which carried the expression cassette for both the Α and β subunits were easily selected on the high selection pressure of the antibiotic Zeocin. The amounts of buTSH recovered were 5-7 mg/L compared to other reported yields 10 mg/L of porcine FSH and [38] 12-16 mg/L of hCG [40]. However, one good observation was that in contrast to some other proteins produced in P. pastoris [8], recombinant buTSH was apparently not degraded as it was purified and apparent molecular weight seen on the SDS-PAGE was comparable too. Importantly there combinant buTSH was not also highly glycosylated as was evident from the molecular weight and also the biological activity.
Interestingly, the reported yield of β subunit in a heterodimeric hormone is much greater than the yield of isolated β subunit. Earlier studies using the CHO cell line also showed that, when the expression of both subunits occurs in the same cell, co-expressed Α subunit assists in the folding of the β subunit, leading to a greater level of recovery of secreted heterodimeric hormone [41]. Therefore, the plausible explanation for good yields (5-7 mg/L) of buTSH might be that the co-expression of Α with β leads to 'rescue' of the β subunit during its folding pathway, leading to secretion of heterodimeric TSH.
The Pichia pastoris expressed hormone is expected to have a glycosylation pattern different from that of native TSH. The Pichia cells do not add sialic acid residues to the carbohydrate moieties on the expressed polypeptide chain but adds a large number of mannose residues. The exact biological significance of the oligosaccharide chains in determining the activity of the glycoprotein hormones is not fully understood but it is known to affect the metabolic clearance rate and biological activity. Hence, the in vivo biological activity is affected by differential glycosylation. Different glycosylation than the native hormone may be one of the reasons that Pichia expressed buTSH was low in bioactivity and had specific activity of only 9 IU/ mg of the purified protein. It is extremely interesting to note that both the native pure buTSH and recombinant buTSH have identical potency in HEK cell cAMP assay i.e. 7-8 IU/mg. This bioactivity is however lower when compared to the purified native bTSH which has 30 IU/mg. The bioactivity also changes when the system of expression is changes as has been seen in case of hTSH where recombinant hormone expressed in CHO cells is more bioactive than the one expressed in Cos 7 cells [42]. In addition the standard for comparison was a native hormone rather than a recombinant hormone. To further improve the bioactivity of the recombinant hormone it needs to be purified in large amounts to study the folding and the CD-ORD spectra.
The successful expression of glycoprotein hormones using the Pichia pastoris expression system has several important implications. In shake flask, after 72 h induction, the secreted clone, 1, secreted up to 7 mg of recombinant buTSH. Interestingly, as the cell density obtained under shake flask conditions is almost 3 times lower than that attained in a fermenter, we can expect to increase the production of recombinant TSH in future when this is done in a fermenter. Thus, by maintaining the per cell productivity constant, it should be possible to achieve almost a 20-fold increase in the yield of the recombinant hormone in a fermenter [8]. Taking these factors into consideration, the Pichia pastoris expression system can be utilized effectively for hyperexpression of properly folded biologically active buTSH for structure-function relationship and therapeutic studies. This system may prove to be a viable system for both the recombinant expression of the glycoprotein hormone subunits and for the study of their folding during biosynthesis. Furthermore, as the Pichia pastoris system has been shown to be suitable for expression of both single and dual radio-labeled proteins (carbon and nitrogen) for NMR studies [43], this system also has the potential of producing adequate amounts of mass isotope labeled hormones and its isolated subunits for structural studies [44,45].
Conclusion
In conclusion, we have shown for the first time that Pichia pastoris was able to produce biologically active buffalo TSH. Recombinant buTSH was probably very similar to its pituitary-derived counterpart at least with respect to its immunological identity and in vitro biological function. However the specific biopotency (i.e. IU/mg protein) is also less in our product when compared to the pituitary derived native bTSH as reported earlier.
Acknowledgement
Work reported in this paper was supported by funds from Department of Biotechnology to RD and KM, from CSIR, Government of India to KM and to NV in the form of Senior Research Fellowship (SRF) and from JC Bose National Fellowship grant of DST, Government of India to KM. We are grateful for this financial support.
References
- Bousfield GR, Perry WM, Ward DN. Gonadotropins: chemistry and biosynthesis. 2nd edn. Knobil E, Neill JD, editors. In: The Physiology of Reproduction. Raven Press Ltd., New York. 1994; 1749-1792.
- Lunenfeld B. Historical perspectives in gonadotrophin therapy. Hum Reprod Update. 2004; 10: 453-467.
- Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. 1981; 50: 465-495.
- Hakola K, Boogaart PV, Mulders J, de Leeuw R, Schoonen W, Heyst JV, et al. Recombinant rat luteinizing hormone: by Chinese hamster ovary cells, purification and functional characterization. Mol. Cell. Endocrinol.1997; 128: 47-56.
- Ludwig M, Doody KJ, Doody KM. Use of recombinant human chorionic gonadotropin in ovulation induction. Fertil Steril. 2003; 79: 1051-1059.
- Caglar GS, Asimakopoulos B, Nikolettos N, Diedrich K, Al-Hasani S. Recombinant LH in ovarian stimulation. Reprod Biomed Online. 2005; 10: 774-785.
- Tarlatzis B, Tavmergen E, Szamatowicz M, Barash A, Amit A, Levitas E, et al. The use of recombinant human LH (lutropin alfa) in the late stimulation phase of assisted reproduction cycles: a double-blind, randomized, prospective study. Hum Reprod. 2006; 21: 90-94.
- Clare JJ, Romanos MA, Rayment FB, Rowedder JE, Smith MA, Payne MM, et al. Production of mouse epidermal growth factor in yeast: high level secretion using Pichia pastoris strains containing multiple gene copies. Gene. 1991; 105: 205-212.
- Cregg JM, Barringer KJ, Hessler AY, Madden KR. Pichia pastoris as a host system for transformations. Mol Cell Biol. 1985; 5: 3376-3385.
- Samaddar M, Catterall JF, Dighe RR. Expression of biologically active beta subunit of bovine follicle-stimulating hormone in the methylotrophic yeast Pichia pastoris. Protein Expr Purif. 1997; 10: 345-355.
- Sen Gupta C, Dighe RR. Hyperexpression of biologically active human chorionic gonadotropin using the methylotropic yeast, Pichia pastoris. J Mol Endocrinol. 1999; 22: 273-283.
- Gadkari R, Deshpande R, Dighe RR. Hyperexpression and purification of biologically active human luteinizing hormone and human chorionic gonadotropin using the methylotropic yeast, Pichia pastoris. Protein ExprPurif. 2003; 32: 175-184.
- Arora T, Muralidhar K. Microheterogeneity in buffalo thyroid stimulating hormone. J. Endocrinol. Reprod. 1999; 3: 70-80.
- Taruna Arora, Nidhi Vashistha, Muralidhar K. Chromatographic Separation of Thyroid Stimulating Hormone from Luteinizing Hormone. Journal of liquid chromatography. 2010; 33: 680-692.
- Sambrook J, Russel G, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual, 2nd edn, New York: Cold Spring Harbor Laboratory Press. 2001.
- Dighe RR, Murthy GS, Kurkalli BS, Moudgal NR. Conformation of the alpha-subunit of glycoprotein hormones: a study using polyclonal and monoclonal antibodies. Mol Cell Endocrinol. 1990; 72: 63-70.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227: 680-685.
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979; 76: 4350-4354.
- Chaudhary R, Jain S, Gupta MN, Muralidhar K. Purification of bubaline luteinizing hormone by gel filtration chromatography in the presence of blue dextran. Process Biochemistry. 2006; 4: 562-566.
- Ulloa-Aguirre A, Midgley AR Jr, Benitins IZ, Padmanabhan V. Immunological and biological activities of hCG following progressive desialylation. Endocr. Rev. 1995; 16: 765-787.
- Albanese C, Christin-Maitre S, Sluss PM, Crowley WF, Jameson JL. Development of a bioassay for FSH using a recombinant human FSH receptor and a cAMP responsive luciferase reporter gene. Mol Cell Endocrinol. 1994; 101: 211-219.
- Sairam MR. Deglycosylation of ovine pituitary lutropin subunits: effects on subunit interaction and hormone activity. Arch Biochem Biophys. 1980; 204: 199-206.
- Chen HC, Shimohigashi Y, Dufau ML, Catt KJ. Characterization and biological properties of chemically deglycosylated human chorionic gonadotropin. Role of carbohydrate moieties in adenylate cyclase activation. J Biol Chem. 1982; 257: 14446-14452.
- Manjunath P, Sairam MR, Sairam J. Studies on pituitary follitropin. X. Biochemical, receptor binding and immunological properties of deglycosylated ovine hormone. Mol Cell Endocrinol. 1982; 28: 125-138.
- Thotakura NR, Bahl OP. Role of carbohydrate in human chorionic gonadotropin: deglycosylation uncouples hormone receptor complex and adenylatecyclase system. Biochem. Biophys. Res. Comm.1982; 108: 399-405.
- Kalyan NK, Bahl OP. Role of carbohydrate in human chorionic gonadotropin. Effect of deglycosylation on the subunit interaction and on its in vitro and in vivo biological properties. J Biol Chem. 1983; 258: 67-74.
- Strickland TW, Pierce JG. The beta subunits of glycoprotein hormones. Formation of three-dimensional structure during cell-free biosynthesis of lutropin-beta. J Biol Chem. 1985; 260: 5816-5819.
- Strickland TW, Thomason AR, Nilson JH, Pierce JG. The common a subunit of bovine glycoprotein hormones: limited formation of the native structure by the totally nonglycosylated polypeptide chain. J. Cell. Biochem. 1985; 29: 225-237.
- Feng W, Huth JR, Norton SE, Ruddon RW. Asparagine-linked oligosaccharides facilitate human chorionic gonadotropin beta-subunit folding but not assembly of prefolded beta with alpha. Endocrinology. 1995; 136: 52-61.
- Ruddon RW, Krzesicki RF, Norton SE, Beebe JS, Peters B, Perini F. Detection of a glycosylated, incompletely folded form of chorionic gonadotropin β subunit that is a precursor of hormone assembly in trophoblast cells. J. Biol. Chem. 1987; 262: 12533-12540.
- Ruddon RW. Super hormones. Nat Biotechnol. 1996; 14: 1224.
- Bousfield GR, Li J, Ward DN. Gonadotropins: chemistry and biosynthesis. In: Knobil and Neill's The Physiology of Reproduction, 3rd edn. Neill JD, editor. Elsevier. 2006; 1581-1634.
- Huth JR, Mountjoy K, Perini F, Ruddon RW. Intracellular folding pathway of human chorionic gonadotropin β subunit. J. Biol. Chem.1992; 267: 8870-8879.
- Huth JR, Perini F, Lockridge O, Bedows E, Ruddon RW. Protein folding and assembly in vitro parallel intracellular folding and assembly. Catalysis of folding and assembly of the human chorionic gonadotropin alpha beta dimer by protein disulfide isomerase. J Biol Chem. 1993; 268: 16472-16482.
- Feng W, Bedows E, Norton SE, Ruddon RW. Novel covalent chaperone complexes associated with human chorionic gonadotropin beta subunit folding intermediates. J Biol Chem. 1996; 271: 18543-18548.
- Huth JR, Norton SE, Lockridge O, Shikone T, Hsueh AJ, Ruddon RW. Bacterial expression and in vitro folding of the beta-subunit of human chorionic gonadotropin (hCG beta) and functional assembly of recombinant hCG beta with hCG alpha. Endocrinology. 1994; 135: 911-918.
- Sudbery PE. The expression of recombinant proteins in yeasts. Curr Opin Biotechnol. 1996; 7: 517-524.
- Richard F, Robert P, Remy JJ, Martinat N, Bidart JM, Salesse R, et al. High-level secretion of biologically active recombinant porcine follicle-stimulating hormone by the methylotropic yeast Pichia pastoris. Biochem.Biophys.Res.Comm. 1998; 245: 847-852.
- Gadkari RA, Sandhya S, Sowdhamini R, Dighe RR. The antigen binding sites of various hCG monoclonal antibodies show homology to different domains of LH receptor. Mol Cell Endocrinol. 2007; 260-262: 23-32.
- Sen Gupta C, Dighe RR. Biological activity of single chain chorionic gonadotropin, hCGalphabeta, is decreased upon deletion of five carboxyl terminal amino acids of the alpha subunit without affecting its receptor binding. J. Mol. Endocrinol. 2000; 24; 157-164.
- Corless CL, Matzuk MM, Ramabhadran TV, Krichevsky A, Boime I. Gonadotropin beta subunits determine the rate of assembly and the oligosaccharide processing of hormone dimer in transfected cells. J Cell Biol. 1987; 104: 1173-1181.
- Joshi L, Murata Y, Wondisford FE, Szkudlinski MW, Desai R, Weintraub BD. Recombinant thyrotropin containing a P-subunit chimera with the human chorionic gonadotropin-P carboxy terminus is biologically active, with a prolonged half life: role of carbohydrate in bioactivity and metabolic clearance. Endocrinology. 1995; 136: 3839-3848.
- Laroche Y, Storme V, De Meutter J, Messens J, Lauwereys M. High-level secretion and very efficient isotopic labeling of tick anticoagulant peptide (TAP) expressed in the methylotrophic yeast, Pichia pastoris. Biotechnology (N Y). 1994; 12: 1119-1124.
- Lustbader JW, Birken S, Pollak S, Pound A, Chait BT, Mirza UA, et al. Expression of human chorionic gonadotropin uniformly labelled with NMR isotopes in Chinese hamster ovary cells: an advance toward rapid determination of glycoprotein structures. Journal of Biomolecular NMR. 1996; 7: 295-304.
- Weller CT, Lustbader J, Seshadri K, Brown JM, Chadwick CA, Kolthoff CE, et al. Structural and conformational analysis of glycan moieties in situ on isotopically 13C, 15N-enriched human chorionic gonadotropin. Biochemistry. 1996; 35: 8815-8823.