
Research Article
Austin J Biotechnol Bioeng. 2014;1(4): 7.
Isolation and Expression of a Chitinase Family Protein AT4G01700 from Arabidopsis Thaliana
Singh A*
Centre of biotechnology, Siksha O Anushandhan University, India
*Corresponding author: Singh. A, Centre of biotechnology, Siksha O Anushandhan University, kalinga Nagar, Ghatikia, Bhubhaneshwar, Odisha, 751003, India.
Received: August 25, 2014; Accepted: September 25, 2014; Published: September 27, 2014
Abstract
Chitinase acts on the chitin present on the cell wall of the fungal pathogen and hence, identification and characterization of the putative gene and the protein is very useful in understanding its role in plant defence. Chitinase gene AT4G01700 was isolated, characterized and expressed from Arabidopsis thaliana (ecotype Arabidopsis Columbia). We wanted to characterize a putative chitinases of Arabidopsis, AT4G01700 which resembled most with the chitinase of Nicotiana tobaccum. The gene was amplified using the cDNA of the plant Arabidopsis thaliana, cloned and expressed using PET EK/LIC vector in different hosts like BL21 (DE3), BL21-pLysS, Rosetta and Rosetta DE3 into a protein up to the size of 31kd. Information obtained from further characterisation of this protein and of chitinase gene (AT4G01700) which the group is continuing with, can be used to develop the transgenic plants resistant to the diseases caused by the fungal pathogens.
Keywords: Chitinases; Arabidopsis thaliana; TAIR database; Pet46EK/LIC vector; Chitinase gene; AT4G01700
Abbreviations
Amp: Ampicillin; CaCl2: Calcium Chloride; cDNA: Complementary DNA; CTAB: Hexadecyltrimethylammonium Bromide; EDTA: Ethylenediamine Tetra Acetic Acid; IPTG: Isopropyl β-D-1-Thiogalactopyranoside; LB: Luria-Bertani; NaOH: Sodium Hydroxide; N-terminus: Amino Terminus; ORF: Open Reading Frame; SDS: Sodium Dodecyl Sulphate; Spp: Species (Plural); TBE buffer: Tris-Borate-EDTA Buffer; β-ME ; Beta Mercaptoethanol; λ; l lambda; μL: Micro Litre; μM: Micro Molar
Introduction
Plants are known to produce various proteins like chitinase in response to the pathogens instead of having their own immune system [1]. Chitinases (EC 3.2.1.14) are the enzymes that catalyze the hydrolysis of the β-1, 4-N-acetyl-D-glucosaaminidic linkages of the polysaccharide chitin, [2] found in the cell wall of most fungi, some algae and in the exoskeleton of arthropods and crustaceans. Chitinase plays different role in different organisms like for growth and molting by insects, for availing carbon and energy source by bacteria. Higher plants and animals produce chitinase to defend themselves from infection of pathogenic fungi [1,2].
"Plant chitinase have both scientific and economic interest". Mostly the plant chitinase are endo-type chitinase which randomly hydrolyze the internal β-1, 4-N-acetyl-D-glucosaminidic linkages of chitin producing chitooligosaccharides. Thus plant chitinase genes can be used to develop the disease resistant transgenic plants that show the increased resistance to the fungal pathogens due to the degradation of chitin present on fungal cell wall by chitinase genes [3,4]. There are data showing the up-regulation of chitinase in response to fungal elicitors, ethylene and other stress [5,6]. Apart from the antifungal activity, chitinase also play important role towards regulation of normal plant development and regulation of legume response to rhizobial nod factors [7,8]. Chitinase are usually involved in active or passive defence against pathogens and they are also known to regulate growth and development by generating or degrading signal molecule and through Programmed Cell Death (PCD).
Plant chitinases are generally the monomeric proteins between 22 and 40 kDa in size. Chitinases have been divided into six classes on the basis of their amino acid sequences [9-11]. Generally plants synthesize a number of closely related chitinase which are encoded by a gene family and further, these enzymes are divided into two classes based upon their acidic or basic isoelectric points [12-14]. Various in vitro studies have demonstrated a growth inhibitory effect of chitinase against fungi containing chitin in the cell wall [15]. Researchers have primarily focused on chitinases from crop plants and ornamental plants [10,16,17]. Although chitinases have been purified and characterized from a variety of sources, very little is known about the enzyme from Arabidopsis thaliana [1]. Simple genome organization and short life cycle have made it a model organism in plant research and offers important advantages for basic research in genetic and molecular biology, the small size of its genome makes Arabidopsis thaliana useful for genetic mapping and sequencing with about 157 mega base pairs and fine chromosomes, gene encoding chitinase have also been used to genetically engineered plants to enhance their protection against fungal pathogens [18]. A. thaliana on the other hand produces a single basic chitinase, encoded by single copy gene [1,19]. In spite of being known as model organism, a very less work has been reported from A.thaliana plant. Since 1991, till date there has been one report on purification and characterization of chitinase from A.thaliana. Chitinase gene AT4G01700 [1]. Chitinase gene AT4G01700 expresses a chitinase family protein. It is involved in carbohydrate metabolic process, cell wall macromolecule catabolic process, it is mainly located in the cell wall and is expressed in 18 plant structure; this gene is expressed during 13 growth stages; it contains the Interpro domain; glycoside hydrolase, family 19 (Interpro:1PR016283); Glycoside hydrolase family 19, catalytic (Interpro 1 PR000726).
In our experiment we focused on Arabidopsis thaliana for the isolation and expression of an uncharacterized chitinase gene AT4G01700 from A. thaliana which may serve an important role in plant defence and thus in making successful transgenic plant.
Materials and Methods
Seed sterilization and plant culture
Seeds of Arabidopsis thaliana (ecotype Columbia) were obtained from IARI, New Delhi. Seeds were taken in separate autoclaved eppendrof tubes and suspended in 70% ethanol for 5 min and then treated with 50% bleach (v/v), 0.1% Tween 20 for 10min. Seeds were resuspended from time to time. Bleach was discarded and seeds were washed thoroughly with a large amount of sterile distilled water. Water was then discarded and the same step was repeated for more than 3 times to remove all bleach from the seeds. Seeds were inoculated in the MS-media (Himedia) plates supplemented with 2% sucrose and wrapped the plates in aluminium foil and were stored at 4°C temperature in dark for 3 days. After 3 days plates were taken out and were kept at room temperature in plant tissue culture room for germination of seeds.
RNA-isolation from the Arabidopsis thaliana seedlings
RNA isolation was done from the seedlings of the Arabidopsis thaliana seedlings (one week old) using RNEASY Minikit (QIAGEN), according to the manufacturer's instructions RLT buffer was used to extract the RNA from the plant tissues. The quality of the RNA isolated was visualized on 1% agarose gel and quantified on nanodrop (Thermo).
c-DNA synthesis and genomic DNA isolation
c-DNA was prepared from the total RNA according to the manufacturer's instructions using the Omni script Reverse Transcription kit (QUIAGEN). In the reaction mixture we took 10X buffer RT 2μL, DNTP 2μl, Oligo dT primer 2μl, RTase 1μl, water 11μl, RNA 2μl, and finally the total reaction mixture was 20μl. The reaction mixture was mixed thoroughly and carefully by vortexing for not more than 5 sec and incubated the mixture at 37°C for 60 min. Genomic DNA was isolated from the fresh seedlings of Arabidopsis thaliana following the instructions on CTAB-method. The genomic DNA was further quantified using the nanodrop (THERMO) and visualized on 1% agarose gel.
Identification of uncharacterized genes of chitinases
As per the protocol published by Xu et al, 2007 (Chitinases in oryza sativa spp. japonica and Arabidopsis spp. ) and Doxey et al, 2007, we separated out different chitinases on the basis of the information given in TAIR data base and using BLAST tool we have short listed AT4G01700, AT4G19820, AT1G56680 as uncharacterized gene.
Sequence retrieval and primer designing
Selected genes were analyzed in TAIR data base and finally primers of all these genes were designed. Putative Arabidopsis family 1 members were identified by searching public databases at the National Centre for Biotechnology information (https://www.ncbi. nlm.nih.gov/BLAST/) and The Arabidopsis Information Resource (https://www.Arabidopsis.org/Blast/). The genome sequence of selected β-1, 3 glucanase genes and the selected c DNA region was taken and pasted the 24-28 base pair sequence from start codon in oligodt analyser and sigma primer calculator. Checked their primer length, GC content, dimer formation and secondary structure.
Amplification of the desired gene
PCR was done using 2μl of c-DNA as template respectively, using the Hot star Hifidelity polymerase (QUIAGEN) and primers for different cycles with modification in annealing temperature and denaturation time using primer of AT4g01700 gene.
Gel extraction and purification
50μl of the amplified PCR product was loaded on 0.7% agarose gel and the part of gel containing the amplified PCR product was cut under the uv-transilluminator. Further the gel-purification was done following the instruction manual of QIA quick GEL Extraction kit (QIAGEN). The gel purified was then stored at -40°C temperature.
Transformation and cloning of AT4g01700 gene
The ligation reaction was performed by using NOVAGEN kit which performed the directional cloning without restriction digestion; it contained PET 46 EK/LIC vector for ligation with desired gene, after performing the ligation and annealing of the desired gene with vector it was transformed in competent cell of DH5α and the colonies were observed on the next day. Similarly transformation was also done using BL21DE3, Rosetta, Rosetta DE3, BL21plysS competent cells. Plating was then done on plates containing antibiotics accordingly- BL21DE3 on LB amp plate, Rosetta DE3 on LB chlor+amp plate, BL21-plysS on LB amp plate and incubated overnight at 37°C temperature.
Plasmid Isolation
Plasmid from DH5α culture was isolated following the instructions of MINI-PREP method of plasmid isolation.
Restriction digestion
Restriction digestion was done using the Hind III enzyme (NEB). Hind III was used as it's the only single cutter enzyme present in the gene AT4G01700 with no other cutting site within the PET EK/ LIC vector, so it appears linear when visualized in 1% agarose gel. The reaction mixture contained 8.0μl of 10X buffer II, 2.0μl Hind III and 58.0μl nuclease free water. 17μl of the master mix and 3μl of the plasmid DNA was added to the each 0.5ml of 4 eppendorf tubes and in other 1.5ml eppendorf tube 58.0μl of the nuclease free water, 2.0μl of the HIND III and 8μl of 10X buffer NEB was taken. Divided equally i.e. 17μl to the 4 separate 0.5ml autoclaved eppendr of tubes each and 3μl of the plasmid DNA was added. For uncut mixture two fresh 0.5ml eppendorf tubes with 17μl of the nuclease free water, 3μl of the plasmid DNA were taken and incubated for 5-6 hrs at 37°C. Visualized the digested sample on an agarose gel.
Optimisation for efficient transformation
The plasmid having the insert gene was used to transform BL21 (DE3) competent cells (Novagen), Rosetta competent cells DE3, BL- 21 pLysS competent cells. In competent cells (100μl) 2 μl plasmid was added with gene of interest. Incubation on ice was given for 5 min. Heat shock was given at 42°C for 45 sec. Again incubated on ice for 2 min. Added 250μl SOC media in each eppendrof and incubated at 37°C for 1 hour and plating was done on the selection media plates having ampicilline and incubated at 37°C overnight. The transformed colonies were observed the next day.
Induction of target protein at different conditions
a) Using IPTG, BL21, DE3 as expression vector: Initially we took 5ml of the LB broth with 2μl of amp in it, and inoculated one colony each from the transformed plate in the LB media and incubated it overnight at 37°C in incubator shaker. Next day 250μl primary culture was inoculated in 10ml LB broth as the OD reaches to 0.4, 1ml culture was taken in a separate eppendorf as uninduced sample (control) and in remaining media added 1mM IPTG to the final concentration of 0.2mM and 1mM and incubated in incubator shaker at 37°C temperature 250 rpm. After one hour of IPTG induction 1ml of the culture was taken and centrifuged at 15000 rpm for 1 min, discarded the supernatant and stored the pellet at -40 ° C temperatures. Similarly pelleted down 1ml IPTG for every 2, 3, 4 and 6 hour and freeze all the samples at -40°C.Visualized on the (12%) SDS gel.
b) Using IPTG, BL21 (pLysS) and Rosetta as a expression vector: The plasmid containing targeted protein was transformed into expression vector BL21 and Rosetta and visualized on 12% SDS gel.
c) Using IPTG, BL21DE3 as expression host with at different temperature, IPTG conc, time: The media was inoculated with transformed BL21 DE3 STRAIN and incubated it overnight at 37°C, 200 rpm. For secondary culture 50ml LB media was taken with 1%glucose (40% stock) and added 1 ml primary culture, incubated it in the incubator shaker at 200rpm, 37 ° C temperatures. After the OD reaches to 0.6, 1ml of the culture was taken in an autoclaved eppendorf for in induced sample and to the rest culture induced with the two different concentration of IPTG a) 0.2 mM IPTG b) 1mM IPTG.
And induced the culture at two different temperature i.e. 18°C and 37°C for both genes, for 2, 4, 6, 8 and 24 hour. 1ml culture was taken after the given interval of time, pelleted down at full rpm for 1 min and stored at -40°C temperature and visualised on 12% SDS gel.
Analysis of proteins on SDS PAGE gel (12%)
The samples were taken out from -40°C and added 45μl of the sample buffer already kept at -40°C and mixed the pellet well to the sample. The buffer was added and boiled at 100°C for 10min. 10μl of the boiled samples was loaded in the gel (12% PAGE) along with 5μl of the protein marker and run. After 2-3 hours when the samples are completely run on gel, the gel was took out and was put on the staining solution (as mentioned above) for 30 min in the rocker, after 30 min decant the staining solution and put the gel in the distaining solution for 5-10 min in the rocker. After 5-10 min decant the distaining solution and added the autoclaved distilled water to the gel and visualized.
Results
Seedling germination and growth
Below are the images showing (A) - showing plate just come out after the overnight incubation at 4°C and (B) showing seedlings germination after 15 days of the incubation. Finally we got seedlings ready for RNA isolation after 15 days of the incubation (Figure 1).
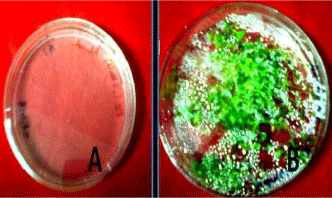
Seedlings of Arabidopsis growing on MS agar plates.
Plate A: Showing the seeds came out after overnight 4°C incubation.
Plate B: Showing the 15 days old grown seedlings.
Figure 1:Seedlings of Arabidopsis growing on MS agar plates.
Plate A: Showing the seeds came out after overnight 4°C incubation.
Plate B: Showing the 15 days old grown seedlings.
RNA Isolation
From the fresh seedlings of Arabidopsis thaliana we isolated the m-RNA using RN easy Minikit (QIAGEN). We obtained two bands of 28S and 18S m-RNA which is shown in the gel image below (Figure 2).
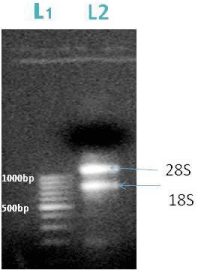
Figure 2: L1-DNA ladder, L2-m RNA; the arrow represents the position of 28S (1000bp) and 18S m-RNA (800bp).
Genomic DNA Isolation
After the mRNA isolation, genomic DNA was isolated from the seedlings of Arabidopsis thaliana plant using CTAB method and visualized on 1% agarose gel. Genomic DNA band were seen in the two lanes.
Amplification of c DNA and genomic DNA using primer of AT4G01700 Gene
For amplification of c DNA and genomic DNA, in the PCR reaction the annealing temperature (55°C) was changed and genomic DNA was taken as template, normal taq polymerase was taken instead of the hot start and visualized on 1% agarose gel. We got band of the amplified gene.
After getting success with PCR on genomic DNA we modified conditions for c- DNA for this we used Hi Fidelity Hot start DNA Polymerase for increasing specificity with modification like addition of Q solution and MgSO4 in reaction mixture. We got an amplified product of about 843 bp (Figure 3).
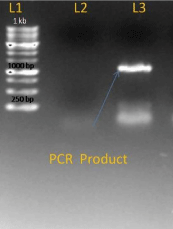
Figure 3: L1-I kb ladder, L2-Negative control, L3-PCR-product.
Gel elution and purification of PCR product of AT4G01700
After conformation of product size the product was eluted with the help of sharp blade under UV transilluminator, from 0.7% agarose gel and finally purified the product from gel by gel elution kit (QIAGEN). After elution the PCR product was purified and was finally visualized on agarose gel (1%). Purified PCR product of the gene AT4G01700 is shown in lane L1 (Figure 4).

Figure 4: L1-1kb DNA Ladder L2-purify PCR product of AT4G01700 Gene.
Ligation and transformation
After the gel elution and purification of the PCR product was done thereafter the ligation reaction was performed by using NOVAGEN kit which performed directional cloning without restriction digestion, it contained PET46 EK/LIC vector for ligation with desired gene, after performing the ligation and annealing of the desired gene with vector it was transformed in competent cell of DH5α and colonies were observed. We got the number of transformed colonies of strain DH5α (Figure 5).
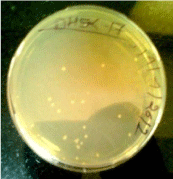
Figure 5: Transformed colony of DH5α.
Plasmid isolation and restriction digestion
In the next step in order to confirm the positive colony, we isolated the plasmid from each transformed colony of gene AT4G01700 using Mini-Prep method. Further on running in gel we visualized the bands showing the plasmid isolated (Figure 6).
Figure 6: L1-1Kb ladder, L2-AT4G01700 (a) plasmid, L3-AT4G01700 (b) plasmid L4-AT4G01700(c) plasmid.
After the plasmid isolation, restriction digestion was performed to confirm the ligation of the insert with the vector. In this we used HindIII restriction enzyme because Hind III is the only single cutter enzyme present in AT4G01700 and it has no other cutting site within the pET EK/LIC vector and so it become linear when visualized on the 1% agarose gel (Figure 7).
Figure 7: L1-1 kb DNA ladder; L2-AT4G01700 (a) insert having no cutting site for Hind III (uncut); L3- AT4G01700(b) insert having no cutting site for Hind III( uncut) L4-AT4G01700(a) insert have single cutting site ; L5-AT4G01700(b) insert have single cutting site.
Next after the confirmation of the ligation of insert with vector, we transformed the plasmid containing insert in to competent cells of BL21 (DE) and many other expression hosts like Rosetta, Rosetta (DE3),BL21(plysS).The resulting transformed colonies are shown below in (Figure 8).
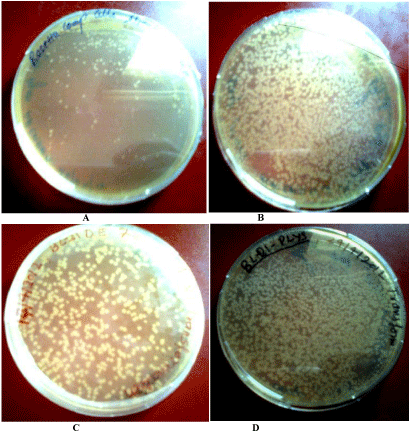
Transformed colonies of Rosetta.
(B): Transformed colonies of rosetta DE3.
(C): Transformed colonies of BL21 DE3.
(D): Transformed colonies of BL21 plys S.
Figure 8(A):Transformed colonies of Rosetta.
(B): Transformed colonies of rosetta DE3.
(C): Transformed colonies of BL21 DE3.
(D): Transformed colonies of BL21 plys S.
Further for the confirmation of the transformation we again isolated the plasmid following the Mini-Prep method and performed the restriction digestion by using HindIII enzyme. (Figure 9); showing restriction digestion with Hind III enzyme in which AT4G01700 gene insert have single cutting site for HindIII enzyme, which becomes linear after digestion.
Figure 9: L1-1 kb DNA ladder L2-uncut (a)no cutting site for HindIII L3- uncut(b) no cutting site for HindIII L4-AT4G01700 (a)gene insert having single cutting site L5- AT4G01700(b) insert having single cutting site.
Expression of targeted protein using IPTG
For the expression of the targeted protein we used the different expression host like BL21 (DE3), BL21pLysS, Rosetta, Rosetta DE3. We also changed the different IPTG concentration at different OD and also gave temperature variation like 18°C and 37°C with IPTG concentration 0.2 to 1. The cells were also induced at lower temperature as has been for expressing Arabidopsis chitinase V (Ohnuma et al; 2011). To prevent the leaky expression we used 0.5% glucose in primary culture and 1% glucose in induced culture. In first experiment we used BL21 (DE3) as expression host. In this we induced with IPTG, when O.D. of the secondary culture reached 0.6. Started the sample collection after each hour until 8 hour, immediately centrifuged the sample and freezed the pellet at -40°C. Resuspended the sample with sample buffer and boiled for 10 min and loaded on 12% SDS gel in SDS gel unit (Bio-Rad) (Figure 10).
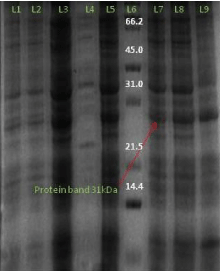
Figure 10: SDS gel image (12%) AT4G01700 gene protein expression of (BL21DE3) L1- 1 hour induced L2- uninduced sample of 1 hour L3- induced sample of 2 hour L4- uninduced sample of 4 hour, L5- induced sample of 4 hour, L6- protein marker, L7- induced sample of 6 hour L8- induced sample of 8 hour L9- induced sample of 10 hour.
Similarly on the expression of the AT4G01700 protein in Rosetta expression host with 0.2 mM IPTG concentration at temperature 18°C temperature with 1% glucose in induced culture we obtained 31 kd of the protein.
And on expressing the AT4G01700 gene protein in plysS expression host and Rosetta (DE3) with 1mM IPTG and 0.2mM concentration respectively at temperature 18°C temperature we again obtained the protein of 31kd. Similarly for the expression of the AT4G01700 gene protein in Rosetta (DE3) expression host with 0.2mM and 1mM IPTG concentration at temperature 37°C and 18°C temperature we again obtained the protein of size 31 kd (Figure 11).

Figure 11: L1- protein marker L2- uninduced sample of 2 hour L3- induced sample of 2 hour L4- induced sample of 4 hour L5- uninduced sample of 4 hour L6- induced 6 hour L7- induced sample of 8 hour L8- induced sample of 24 hour L9- uninduced sample of 24 hour.
Discussion
In the experiment the uncharacterized gene AT4G01700 of the model plant Arabidopsis thaliana was isolated, characterized and cloned, further we are trying to express the protein using various optimization in temperature, IPTG concentration etc. Today fungal pathogens cause huge loss to the crop plants and chitinase being an antifungal protein acts on the chitin present in the exoskeleton of the fungal pathogens hence considered suitable for the management of the pests and pathogens. The gene of our interest AT4G01700 along with the other genes can be characterized and in future can be used for the development of the transgenic plants resistant against the fungal pathogens. Apart from the model plant Arabidopsis thaliana chitinase having antifungal property have also been reported from the other sources. Molecular cloning, characterization and expression of chitinase PjChi-1 from fungus Paecilomyces javanicus which is reported to inhibit the growth of phytopathogenic fungi such as Sclerotium rolfsii, Colletotrichum gloeosporioides, Aspergillus nidulans and Rhizoctonia solani [20]. Chitinase gene has also been isolated and characterized from the carnivorous plant Nepenthes khasiana and the results showed the involvement of gene encoding chitinase in prey-trap interaction, and their differential expression and activity during prey trapping [21]. The gene encoding an extracellular chitinase from marine Alteromonas spp. strain O-7 was cloned in Escherichia coli JM109 by using pUC18 [22]. Apart from this several acidic and basic chitinases have also been isolated from the Arabidopsis thaliana plant [1]. Chitinase is often produced in higher plants as a general defence response after wounding or pathogenic attack. A putative role for chitinase in muskmelon seeds is defence against fungal pathogen [23]. So on the basis of research paper published by Xu etal and Doxey et al, 2007, we separated out different chitinases and on the basis of the information given in TAIR database and using BLAST tool we found the uncharacterized gene AT4G01700, AT1G56680, AT4G19820 and analyzed in TAIR database. Putative Arabidopsis family 1 members were identified by searching public databases at the National Centre for Biotechnology Information (https://www.ncbi. nlm.nih.gov/BLAST/) and The Arabidopsis Information Resource (https://www.Arabidopsis.org/Blast/). In the paper by Verburg et.al, 1991, purification and characterization of basic chitinase from Arabidopsis Thaliana was reported and the enzyme of 32 kilodalton was determined by SDS PAGE. And in the paper by Deborah et al (1990), the genes encoding the acidic and basic chitinase were cloned and the nucleotide sequence was determined and the expression of both acidic and basic chitinases on different plants parts was checked. In our experiment we tried to isolate and express a chitinase family protein AT4G01700 which can prove to act as an antifungal protein useful for the preparation of transgenic plants, gene AT4G01700 was characterized. The gene was amplified using the cDNA of the plant Arabidopsis thaliana (ecotype Arabidopsis Columbia). The gene was cloned and expressed using PET EK/LIC vector in different hosts like BL21 (DE3), BL21-pLysS, and Rosetta DE3. Protein up to 31Kd was expressed and further trying to express the protein using various optimization. The chitinase protein is further targeted to develop the transgenic plants resistant against the fungal pathogens.
Conclusion
The gene AT4G01700 can be further purified and can be used as an antifungal protein to check the pathogen attack on plants, and depending upon the expression level in different parts of the plant we can lead further towards development of transgenic plants through introducing such genes into different plant species susceptible to fungal pathogens.
References
- Verburg JG, Huynh QK. Purification and Characterization of an Antifungal Chitinase from Arabidopsis thaliana. Plant Physiol. 1991; 95: 450-455.
- Punja ZK, Zhang YY. Plant chitinases and their roles in resistance to fungal diseases. J Nematol. 1993; 25: 526-540.
- Brogue K, Chet I, Holliday M, Cressman R, Biddle P, Knowlton S, et al. Transgenic Plants with Enhanced Resistance to the Fungal Pathogen Rhizoctonia solani. Science. 1991; 254: 1194-1197.
- Nishizawa Y, Nishia Z, Nakazono K, Soma M, Nakajima E, Vgaki M, Hibi T. Enhanced resistance to blast (Magnaporthegrisea) in transgenic Japonica rice by constitutive expression of rice chitinase. Theor Appl. Genetics. 1999; 99: 383-390.
- Boller T. Ethylene and the regulation of antifungal hydrolase in plants Survey of Plant. Molecular and cell biology. 1988; 145-174.
- Watanabe A, Nong VH, Zhang D, Arahira M, Yeboah NA, Udaka K, et al. Molecular cloning and ethylene-inducible expression of Chib1 chitinase from soybean (Glycine max (L.) Merr.). Biosci Biotechnol Biochem. 1999; 63: 251-256.
- Zhong R, Kays SJ, Schroeder BP, Ye ZH. Mutation of a chitinase-like gene causes ectopic deposition of lignin, aberrant cell shapes, and overproduction of ethylene. Plant Cell. 2002; 14: 165-179.
- De Jong AJ, Heidstra R, Spaink HP, Hartog MV, Meijer EA, Hendriks T, et al. Rhizobium Lipooligosaccharides Rescue a Carrot Somatic Embryo Mutant. Plant Cell. 1993; 5: 615-620.
- Collinge DB, Kragh KM, Mikkelsen JD, Nielsen KK, Rasmussen U, Vad K. Plant chitinases. Plant J. 1993; 3: 31-40.
- Beintema JJ. Structural features of plant chitinases and chitin-binding proteins. FEBS Lett. 1994; 350: 159-163.
- Meins F, Fritig BJ, linthorst HJM, Mikkelsen JD, Nevhans JM, Ryals J. Plant chitinase gene. Plant Mol.Biol.1994; 12: S22-S28.
- Kombrink E, Schröder M, Hahlbrock K. Several "pathogenesis-related" proteins in potato are ,3-beta-glucanases and chitinases. Proc Natl Acad Sci U S A. 1988; 85: 782-786.
- Legrand M, Kauffmann S, Geoffroy P, Fritig B. Biological function of pathogenesis-related proteins: Four tobacco pathogenesis-related proteins are chitinases. Proc Natl Acad Sci U S A. 1987; 84: 6750-6754.
- Mauch F, Hadwiger LA, Boller T. Antifungal hydrolases in pea tissue I. Purification and characterization of two chitinase and two β-, 3-glucanases differentially regulated during development and in response to fungal infection. Plant physiology. 1988; 87: 325-333.
- Brokert WF, Van Parihs J, Allen AK, Peumans WJ. Comparison of some molecular, enzymatic and antifungal properties of chitinases from thron apple, tobacco and wheat. Physiol. Mol. Plant Pathol.1988; 33: 319-331.
- Meins F, Fritig BJ, linthorst HJM, Mikkelsen JD, Nevhans JM, Ryals J. Plant chitinase gene. Plant Mol.Biol.1994; 12: S22-S28.
- Shinshi H, Neuhas JM, Ryals J, Meins F Jr. Structure of a tobacco endochitinase gene: evidence that different chitinase genes can arise by transposition of sequences encoding a cysteine-rich domain. Plant Mol Biol. 1990; 14: 357-368.
- Meyerowitz EM, Pruitt RE. Arabidopsis thaliana and Plant Molecular Genetics. Science. 1985; 229: 1214-1218.
- Samac DA, Hironaka CM, Yallaly PE, Shah DM. Isolation and Characterization of the Genes Encoding Basic and Acidic Chitinase in Arabidopsis thaliana. Plant Physiol. 1990; 93: 907-914.
- Alexandre de Siqueira Pinto, Cristine CB, Marilene HV, Augusto S, Cirano JU. Purification and characterization of an extracellular chitinase from the entomopathogen Metarhizium anisopleio. Canadian Journal of Microbiology. 1997; 43: 322-327.
- Eilenberg H, Pnini-Cohen S, Schuster S, Movtchan A, Zilberstein A. Isolation and characterization of chitinase genes from pitchers of the carnivorous plant Nepenthes khasiana. J Exp Bot. 2006; 57: 2775-2784.
- Witmer X, Nonogaki H, Beers E P, Bradford K J, Welbaum G E. Characterization of chitinase activity and gene expression in muskmelon seeds. Seed Science Research. 2007; 13:167-178.
- Kaomek M, Mizuno K, Fujimura T, Sriyotha P, Cairns JR. Cloning, expression, and characterization of an antifungal chitinase from Leucaena leucocephala de Wit. Biosci Biotechnol Biochem. 2003; 67: 667-676.