
Editorial
Austin J Biotechnol Bioeng. 2015; 2(4): 1049.
Microbial Genome Sequencing Projects: Foundation for Comparative Genome Analysis
Tatiana Tatusova*
National Center of Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, USA
*Corresponding author: Tatiana Tatusova, National Center of Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894, USA
Received: August 08, 2015; Accepted: August 28, 2015; Published: August 31, 2015
Editorial
Microbes are probably the most abundant and diverse group of cellular organisms on Earth. The number of described species is now about 12,000, and the number of species on earth is estimated in the millions [1]. Bactria can be found living in nearly every habitat on the face of the earth, regardless of how seemingly inhospitable: they have been found in the deepest parts of the ocean, seven miles under the surface and as high as 40 miles into the atmosphere; many species of bacteria can withstand harsh conditions, including extreme heat, cold and saline. Sequenced microbial genomes represent a large collection of strains with different levels of quality and sampling density. They include many important human pathogens, but also organisms that are of interest for non-medical reasons, i.e. biodiversity, epidemiology, ecology. These are obligate intracellular parasites, symbionts, free-living microbes, hyperthermophiles and psychrophiles, and aquatic and terrestrial microbes, all of which have provided a rich insight into evolution and microbial biology and ecology. There is almost 20-fold range of genomes sizes spanning from ultra-small 45 kb archaeal genome of Candidatus Parvarchaeum acidiphilum obtained from mine drainage metagenome project to the largest, so far - 15, 8Mb, recently submitted draft assembly of Mastigocoleustestarum and the largest complete genome (14, 7 Mb) of Sorangiumcellulosum, alkaline-adaptive epothilone producer [2]. The distribution of genome size for all the genomes in public archive is shown on Figure 1. There is a big variation in genome structure: there are organisms with single circular chromosomes, but also organisms with linear chromosomes, multiple chromosomes, and a mixture of chromosomes and extra chromosomal elements including plasmids. The GC-content of bacterial genomes also spans a large range, from extremely low, 13.5%, for the obligate intracellular symbiotic microbe, Zinderiainsecticola, [3] to 74.8% for the facultative anaerobic soil bacterium of Anaeromyxobacter [4].
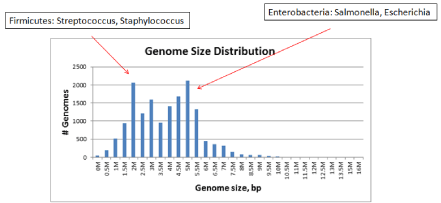
Figure 1: Genome size distribution. The smallest archaeal genome (45 kB)
of Candidatus Parvarchaeum acidiphilum: the largest, so far - 15,8 Mb, draft
assembly of Mastigocoleus testarum and the largest complete genome
(14,7 Mb) of Sorangium cellulosum, alkaline-adaptive epothilone producer.
The picks on the histogram correspond to the most abundant families in
Firmicutes with a typical size of 1,5 Mb and Enterobacteria with typical size
of 5 Mb.
From the beginning of microbial genome sequencing era researchers have shown a commitment to phylogenetic diversity and the completion of one genome from each prokaryotic division or phylum is still a frequently articulated community goal. However, largely because of interest in human pathogens and advances in sequencing technologies, there are also now a number of very closely related genomes whose organization and gene content can be directly compared. Rapidly growing microbial genome data set at the National Center for Biotechnology Information (NCBI) at the time of writing (August 2015) contains almost 45, 000 prokaryotic genome assemblies representing over 7, 000 different species from 52 phyla in Bacteria and 12 phyla in Archaea (including unclassified and ‘candidate’) (see Figure 2 for the genome distribution by major phyla).
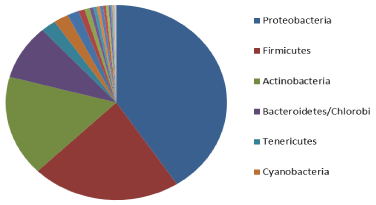
Figure 2: Distribution of bacterial species by phyla. Top six phyla with more
than 100 species sequenced: Proteobacteria – 2291; Firmicutes – 1240;
Actinobacteria – 945; Bacteroidetes/Chlorobi group – 512; Tenericutes – 134;
Cyanobacteria – 103.
Recent rapid advances in sequencing technologies provided a relatively cheap and fast way of studying the diversity of microbial species by discovering representatives of novel divisions or even phyla [5,6] and analyzing the variation within the species by sequencing closely related genomes from the ecological microbial populations or clinical studies of pathogenic bacteria.
Historically, prokaryotic organisms were organized by classical taxonomic ranking system (species, genus, family, order, and phylum). Delineation of prokaryotic species was originally based on phenotypic information, pathogenicity and environmental observations. A recent review [7] describes the history and present state of various methods of description of prokaryotic species. The authors suggest the concept of species as “a category that circumscribes monophyletic, and genomically and phenotypically coherent populations of individuals that can be clearly discriminated from other such entities by means of standardized parameters”. Scientists from different disciplines (taxonomists, ecologists and evolutionary biologists) have different interpretations of species defined by the framework of their needs and the tools they use for identification. For almost 50 years DNADNA Hybridization (DDH) has been the gold standard method for prokaryotic species delineation at the genomic level. New approaches to the identification of microbial species are taking into account the advantages of the growing massive volume of genomic sequence data [8,9]. Several groups have attempted to delineate the taxonomy of Archaea and Bacteria using the methods based on single-copy universally conserved gene markers [10-14]. Other method employ genome sequence directly without pre-annotation of genes. Average Nucleotide Index (ANI) is an silico version of DDH method. JSpecies package [15] provides an interface to calculate ANI for a pair of genomes that can be used for species classification. The ANIbased genome tree can be used to model the organism evolutionary relationships. Genome Blast Distance Phylogeny (GDBP) method [16] is another implementation of in silico DDH method that infers genome-to-genome distances between genome pairs from genome sequences. K-mer based approach uses the number of co-occurring k-mers (substrings of K nucleotide in genomic sequence) as the distance between the pair of genomes that represent the evolutionary relatedness [17]. The benchmarking of genome based classification methods can be found in the recent review [18].
It is now known that intra species variation can be as significant as interspecies diversity. Bacterial genomes from various strains of the same species can vary considerably in genome size, nucleotide composition and gene content. It has become clear that, bacterial species cannot be represented by an individual reference strain or a set of reference genomes. The ‘pan-genome’ concept has been introduced by Tettelin et al. in 2005 [19]. The pan-genome has been defined as a super-set of all genes in all the strains of a species. A pan-genome includes the “core genes” that are present in nearly all strains; “accessory genes” present in two or more strains, and finally “unique genes” specific to single strains. A shift in the paradigm from individual genome to ‘pan-genome’ has occurred in the past few years, with the rapid advances in the sequencing technology. The main approach in pan-genome studies is a comparative analysis of multiple strains from a single species, although one can also describe pan-genomes for different taxonomy levels – for example, a phylum or genus pan-genome, or sometimes even a subspecies pan-genome (as in the case of E. coli O157:H7, with 34 genomes sequenced so far). Alternative approaches include the use of rapidly growing metagenomic sequence data and single-cell genome sequencing. The pangenome concept is already changing the way we understand bacterial evolution, adaptation, and population structure, and has further important implications in identification of virulence genes [20].
A recent shift in the paradigm changes the field of comparative and population genomics. This emerging field leads to the development of new algorithms, statistical models, and visualization tools. It is affecting all areas of bioinformatics including data storage and data management, genome assembly and annotation, protein clustering, phylogenetic trees construction.
Over the last 20 years, the National Center for Biotechnology Information (NCBI), as a primary public repository of genomic sequence data, has been collecting and maintaining humongous amounts of heterogeneous data [21]. The databases vary in size, data types, design and implementation. They cover most of the genomic biology data types including the project description, project sequence data (genomic, transcript, protein sequences), sequence reads and related bibliographical data. All these databases are integrated in a single Entrez system and use a common engine for data search and retrieval. This provides researchers with a common interface and simplifies navigation through the large information space.
The Genome database was first created in 1995 when the complete genome of Haemophilus influenza, the first cellular organism was sequenced at TIGR [22] and submitted to GenBank. The genome data has changed dramatically over the past 20 years of microbial genome sequencing [23]. The newly redesigned Genome resource organizes information about the organism (usually at the species level) and provides the summary view of the data from all “genome-scale” projects: map, genome, assembly, annotation, transcriptome etc. The daily updated list of all genomes can be browsed via web (https://www. ncbi.nlm.nih.gov/genome/browse/) or downloaded via FTP (ftp://ftp. ncbi.nlm.nih.gov/genomes/).
NCBI, as a primary data archive, traditionally collected species and strain designation from the submitters of the sequence data. In the absence of the independent validation methods that leads to potential problems with the taxonomic identification. One of the most premises when classifying new taxa is the designation of one of the strains as being the type material that should be used as reference for any further taxonomic work. NCBI Taxonomy [24] team has started a project to curate type material in the taxonomy, and use it to flag sequence from type in GenBank. NCBI is developing new approaches to the classification of genome sequence and correction of sequence tags in GenBank. They employ all three described above computational methods (marker-based, K-mer, ANI) to construct the genome distance trees and evaluate the species assignment. Sequences from type can use those as landmarks of correct identification to help resolve misidentified genomes.
The data in microbial genomes collection have different levels of sequence and assembly quality; some sequencing technologies have known high level of error rate that lead to the large number of ambiguities and low quality nucleotides in the final consensus sequence. Many genomes assemblies coming from single cell sequencing technology give only partial representation of DNA in a cell, ranging from 10% to 90%. NCBI Refseq project aims to provide a high quality data set of genome, transcript and protein sequence data. The source of the genomic sequence in the Refseq collection is a primary sequence record in the International Nucleotide Sequence Database Consortium, INSDC, public archives [25].All assemblies with full representation of the genome of a single organism that pass validation quality are taken into Refseq. Genome assembly quality validation criteria are described in details in [26-28]. Genome assemblies from environmental samples, mixed cultures, hybrid organisms and chimeras submitted to GenBank are not accepted into RefSeq because they do not represent a single bacterial organism.
Integrated microbial genome resource provides an infrastructure for comparative genome analysis. Microbial sequencing projects now span from complete and draft genome assemblies of isolated organisms to large-scale comparative genomic projects of multiple strains, and to the new field of metagenomics where the entire complement of DNA from a given ecological niche is being sequenced. The information form tens of thousands of sequenced genomes has already provided an insight into microbial diversity, evolution, and ecology. Advances in sequencing technologies continue to change the field of genomics creating great opportunities of developing new bioinformatics approaches and computational sequence analysis methods.
References
- Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014; 12: 635-645.
- Han K, Li ZF, Peng R, Zhu LP, Zhou T, Wang LG, et al. Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci Rep. 2013; 3: 2101.
- McCutcheon JP, Moran NA. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol. 2010; 2: 708-718.
- Lucas S, Copeland A, Lapidus A, Glavina del Rio T, Dalin E, Tice H, et al. Complete sequence of Anaeromyxobacter sp. K. Unpublished.
- Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013; 499: 431-437.
- Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015; 523: 208-211.
- Rosselló-Móra R, Amann R. Past and future species definitions for Bacteria and Archaea. Syst Appl Microbiol. 2015; 38: 209-216.
- Thompson CC, Amaral GR, Campeão M, Edwards RA, Polz MF, Dutilh BE, et al. Microbial taxonomy in the post-genomic era: rebuilding from scratch? Arch Microbiol. 2015; 197: 359-370.
- Chun J, Rainey FA. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int J Syst Evol Microbiol. 2014; 64: 316-324.
- Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006; 311: 1283-1287.
- Mende DR, Sunagawa S, Zeller G, Bork P. Accurate and universal delineation of prokaryotic species. Nat Methods. 2013; 10: 881-884.
- Lang JM, Darling AE, Eisen JA. Phylogeny of bacterial and archaeal genomes using conserved genes: supertrees and supermatrices. PLoS One. 2013; 8: e62510.
- Wu D, Jospin G, Eisen JA. Systematic identification of gene families for use as "markers" for phylogenetic and phylogeny-driven ecological studies of bacteria and archaea and their major subgroups. PLoS One. 2013; 8: e77033.
- Darling AE, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014; 2: e243.
- Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009; 106: 19126-19131.
- Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013; 14: 60.
- Chan CX, Ragan MA. Next-generation phylogenomics. Biol Direct. 2013; 8: 3.
- Larsen MV, Cosentino S, Lukjancenko O, Saputra D, Rasmussen S, Hasman H, et al. Benchmarking of methods for genomic taxonomy. J Clin Microbiol. 2014; 52: 1529-1539.
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl AcadSci U S A. 2005; 102.
- Vernikos G, Medini D, Riley DR, Tettelin H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015; 23: 148-154.
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015; 43: D6-17.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilusinfluenzae Rd. Science. 1995; 269: 496-512.
- Land M, Hauser L, Jun SR, Nookaew I, Leuze MR, Ahn TH, et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics. 2015; 15: 141-161.
- Federhen S. Type material in the NCBI Taxonomy Database. Nucleic Acids Res. 2015; 43: D1086-1098.
- Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2015; 43: D30-35.
- Tatusova T, Ciufo S, Federhen S, Fedorov B, McVeigh R, O'Neill K, et al. Update on RefSeq microbial genomes resources. Nucleic Acids Res. 2015; 43: D599-605.
- Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 2014; 42: D553-559.
- Land ML, Hyatt D, Jun SR, Kora GH, Hauser LJ, Lukjancenko O, et al. Quality scores for 32,000 genomes. Stand Genomic Sci. 2014; 9: 20.