
Research Article
Austin J Biotechnol Bioeng. 2017; 4(4): 1085.
Molecular Analysis of Hb-E and Beta-Thalassemia Major Patients among Bangladeshi Population
Aziz A1,2*, Ahmed W2, Sarwardi G2, Ara Naznin R2, Rehena J1 and Ferdoushi A1
¹Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Bangladesh
²Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Bangladesh
*Corresponding author: Abdul Aziz, Department of Biochemistry and Molecular Biology, Dhaka Shishu Hospital, Dhaka, Bangladesh
Received: November 10, 2017; Accepted: December 19, 2017; Published: December 26, 2017
Abstract
Background: Beta thalassemia is one of the most common haemoglobin blood disorder in Bangladesh. More than 500 alleles have been characterized in or around the beta-globin region. Different geographical regions show different frequency of allelic characteristics. In this study, we explored the spectrum of Beta-thalassemia (b-thal) alleles followed-up in beta-thalassemia major and Hb E disease.
Methods: One hundred thalassemia major and Hb E beta thalassemia samples were collected patients in attended Dhaka Shishu hospital, Bangladesh. Mutation analysis was performed by amplification refractory mutation system (ARMS-PCR).
Results: A total of eight different beta-thalassemia mutations were characterized among 200 alleles. The most common mutation defected IVS1-5 (G>C) (55.5%), followed by Cd 26 (G>A) (25%), Fr 8-9 (+G) (3.5%), Fr 41-42 (-TTCT) (3%), IVS 1-1 (G>T) (1.5%), Cd 30 (G>C) (1%) and unknown 10.5% in this study.
Conclusion: ARMS–PCR technique is used as simple, rapid and non radioactive method and valuable molecular tool for beta thalassemia and Hb E mutation detection. In future this study help to detect common mutation in Bangladesh that will facilitate the implementations of genetic counselling and prenatal diagnosis in the population of Bangladesh.
Keywords: Beta-thalassemia; ARMS; PCR; Prenatal diagnosis
Introduction
Thalassemia is the most common inherited autosomal recessive gene disorder. It is caused by impaired synthesis of globin chains that alters production of Hemoglobin (Hb) [1,2]. Thalassemia causes varying degrees of anemia, which can range from significant to life threatening. People of Mediterranean, Middle Eastern, African, and Southeast Asian descent are at higher risk of carrying the genes for thalassemia [3]. The prevalent of thalassemia is increasing throughout the world today [4,5]. According to a report by the World Health Organization, 3% of the Bangladeshi populations are carriers of beta-thalassaemia and 4% are carriers of HbE [6]. In tribal school children the prevalence of the HbE trait was 41.1%, while that of betathalassaemia was 4.2 percent [7]. Being a problem at the genetic level, the disease cannot be cured completely and hence the current trend to cope with the disease is ‘‘prevention’’ rather than ‘‘cure’’ [8].
Majority of thalassemia patients are born in countries with limited resources and suffer from infection diseases and malnutrition [9]. Beta-thalassemia major and Hb E-beta-thalassemia patients do not survive more than 5 years without blood transfusion [10]. Betathalassaemia patients need huge amount of donated blood but blood screening is expensive (Figure 1). As a result, thalassemia patients involve with complications, including hepatitis B, hepatitis C and HIV [11].
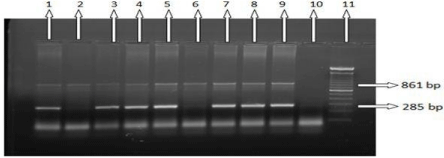
Figure 1: ARMS-PCR and Agarose gel electrophoresis analysis for IVS-1-
5 (G>C) mutation. Common IVS-1-5 (Mt and N) which as reverse ARMS
primers and amplify a 285 bp fragment. Here, Lanes 1, 3, 4, 5, 7, 8, 9: Positive
for IVS-1-5 (G>C) mutation; lanes 2,6: Negative for IVS-1-5 (G>C) mutation;
lane 10: Blank for this mutation; lane 11: 100bp ladder.
Recently, β-thalassemia causes more than 200 mutations have been detected the diverse levels of beta globin gene expression [12]. These mutations are not uniformly distributed, but have a geographical specificity and racial origin, as each is characterized by the presence of few common mutations and variable numbers of rare ones [13]. In Bangladesh most common type of mutation is IVS 1-5 (G>C) and Cd 26 (G>A) [14]. Since 1990, the Amplification Refractory Mutation System (ARMS) has been widely used in β-thalassemia mutation detection with primers that are complementary to the normal and mutant DNA sequences [15].
Materials and Methods
The study consisted of 100 confirmed thalassemia patients. The patients who came to Dhaka Shishu Hospitals for treatment were selected as subjects. Written informed consent was taken from all participants to make the study ethically sound (Figure 2). Peripheral blood was collected into EDTA containing tube. Blood samples from patients were stored in 4°C. DNA was isolated from peripheral blood using PureLink@ Genomic DNA Purification Mini Kit (Gendra, USA). This method relies on phase separation by centrifugation of a mix of the aqueous sample. The kit was used according to the manufacturer’s instructions. Extracted DNAs were tested to determine the purity and concentration of DNA per micro-litter volume through Qubbit® 2.0 Fluorometer.
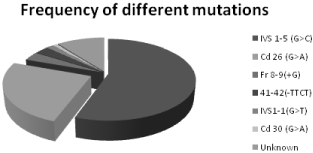
Figure 2: Statically data of different mutations in Bangladeshi population.
ARMS-PCR (Amplification Refractory Mutation System) allows the characterization of point mutations directly by the presence or absence of amplification using allele specific primers [16]. For the diagnosis of specific point mutation a pair of allele-specific primers one of which has its 3' terminal nucleotide complementary to the point mutation (Mt ARMS primer) and other to the normal DNA sequence (N ARMS primer) was used (Table 1). Eight common mutations were chosen for this study (Table 1).
![]()
Primer Name
Sequence(gene specific)
Length (bp)
Amplicon size
IVS1-5(G>C)M
5’-CTCCTTAAACCTGTCTTGTAACCTTGTTAG-3’
30
285
IVS1-5 N
5’-CTCCTTAAACCTGTCTTGTAACCTTGTTAC-3’
Cd 26 (G>A)
5’-TAACCTTGATACCAACCTGCCCAGGGCGTT-3
30
267
Fr8-9(+G) M
5’-CCTTGCCCCACAGGGCAGTAACGGCACACC-3’
30
215
Fr8-9 N
5’-CCTTGCCCCACAGGGCAGTAACGGCACACT-3’
Fr41-42(-TTCT) M
5’-GAGTGGACAGATCCCCAAAGGACTCAACCT-3’
30
439
Fr41-42 N
5’-GAGTGGACAGATCCCCAAAGGACTCAAAGA-3’
Codon 30(G-C) M
5’-TAAACCTGTCTTGTAACCTTGATACCTACG-3’
30
280
Cd 30 N
5’-TAAACCTGTCTTGTAACCTTGATACCTACC-3’
Codon 15(G-A) M
5’-TGAGGAGAAGTCTGCCGTTACTGCCCAGTA-3’
30
500
Cd 15 N
5’-TGAGGAGAAGTCTGCCGTTACTGCCCAGTG-3’
Del 619bp-F
5’-CAATGTATCATGCCTCTTTGCACC-3’
24
242
Del 619BP-R
5’-GAGTCAAGGCTGAGAGATGCAGGA-3’
IVS1-1(G-T)M
5’-TTAAACCTGTCTTGTAACCTTGATACGAAA-3’
30
281
IVS1-1 N
5’-GATGAAGTTGGTGGTGAGGCCCTGGGTAGG-3’
Control
A 5’-CAATGTATCATGCCTCTTTGCACC-3’
24
861
B 5’-GAGTCAAGGCTGAGAGATGCAGGA-3’
Table 1: Primer sets which are selected for ARMS analysis of mutations.
PCR reaction mixture contained genomic DNA (100ng), MgCl2 (50 mM), Buffer (10X mM), DNTPs (10mM), Taq polymerase, normal (N) or mutant (M) primer (10 pM), Internal Control Primer (10 pM) and Deionized water. The reaction tubes were then put on to thermal cycles on a DNA thermal cycler (Bio-Rad, Germany). To detect the mutations the ARMS-PCR was programmed (30 cycles: preheating at 95°C for 3 min, denaturing at 95°C for 20 sec, annealing temperatures 65°C for 45 sec, and extension at 72°C for 1 min and Final extension 72°C for 3 min). The ARMS-PCR products and the ladder marker are resolved by electrophoresis. 10μl of the product are loaded on 2% agarose gel TAE buffer with Ethidium bromide solution and run at 100 volt and 300 atm for 30 min. In addition, bands are visualized on UV transiluminator and then photographed by using photo documentation system. All samples were analyzed with positive and negative controls for particular mutation. An 861bp internal control band was also amplified in all reactions indicating successful PCR. The PCR products were finally analyzed by electrophoresis.
Results and Discussion
In this research plan 100 thalassemic patients were confirmed by High Performance Liquid Chromatography (HPLC). Among 200 allelic mutations our eight primers were covered 179 allelic mutations. IVS1-5(G> C) most appearance in 100 thalassemic patients is 55.5%, and also Cd 26 (A>G) 25%, Fr 8-9(+G) 3.5%, Fr 41-42(-TTCT) 3%, IVS1-1(G>T) 1.5%, Cd 30 (G>A) 1% and unknown 10.5%
The B-globin gene is a relatively small gene (2000bp) located in the short arm of chromosome 11 [17]. Although 200 causative mutations have been reported for B-thalassemia syndromes. The frequencies of mutations in most populations usually consist of a number of common mutations and a slightly number of rare mutations [18].
In this study, two hundred thalassemia allelic statuses were included. IVS1-5 (G>C) (55.5%) is the most common mutation in Bangladesh. Our study is similar with previous analysis of Beta goblin gene among Chittagong population in Bangladesh IVS I-5(G>C) frequency 39.1% [19].
IVS I-5 (G > C) is the most prevalent β-thalassemia mutation in South Asia but the frequency varies ranging from 36.5% in Pakistan, 56.3% in India and to 64.6% in Sri Lanka [20].
The second common mutation was Cd 26 (G>A) that is 25%. In Bangladesh, Hb E disease is most prevalent that mutation occur by Cd 26 (G>A) homozygous and E-beta thalassemia mutation are Cd 26 (G>A) and other beta mutation. 50,000 – 100,000 children with β-thalassaemia major die each year in low and middle income countries. [International thalassemia federation]. The prevalence of Hb E trait in Bengali school children was 6.1%, while in tribal school children it was 41.7% [7]. It is however mostly concentrated on the border between Thailand, Laos and Cambodia an area known as Hb E triangle [21]. In Cambodia, Laos, Myanmar and Thailand, the carrier status of HbE trait were 30%, 35%, 28%, 13% respectively [22]. Hb E in India is most frequently seen in Eastern and Far Eastern part of India (average frequency of 10.9%) with highest frequency seen in Assam, Meghalaya, Arunchal Pradesh, West Bengal, Manipur and Nagaland [23].
Other common mutations were found Fr8-9 (+G), IVS I-1(G>T), Fr 41-42 (-TTCT), Cd 30(G>C) total percentage of 9% and Chittagong with Indian subcontinent 12.5% [19]. In Indian subcontinent has a lot of significance of various mutations that case is migration. For instance, IVS-I-5(G>C) has Asian Indian and Southeast Asian origins [24–26]; codons 41/42 has Chinese, Southeast Asian and Asian Indian origins [25,27]; codons 8/9 and codon 15 (G>A) both have Asian Indian origin [25]. The Bangladeshi population has a very important and vivid history of migration between India and Bangladesh. At first British, Portuguese dominated this subcontinent. In 1947, Indian subcontinent liberated from British and those moment India and Pakistan was separated. Bangladesh was part of Pakistan and Pakistan declared Bangladesh would become Muslim country as a result Hindus was starting to migrate to India as well as Muslim migrated to Bangladesh. Last of all Bangladesh was liberated from Pakistan in 16th December, 1971. This migration means mixture of different alleles from Indian subcontinent, British, Pakistan and Portuguese etc.
Conclusion
Bangladesh is a developing country and has limited resource. Seven different β-thalassemia mutations have been presented in the Bangladeshi population. The most frequent mutations in Bangladesh were IVS 1-5 (G>C), Cd 26 (G>A) and Fr 8-9 (+G). ARMS–PCR technique is used as simple, rapid and non radioactive method and valuable molecular tool for beta thalassemia mutation detection.
Acknowledgement
We thank Dr. A K M Mohiuddin (Professor, Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Bangladesh) for encouragement and inspiration and scientific officer of Dhaka Shishu Hospital for their kindness help, well behavior, valuable suggestions and encouragement to complete the research work.
References
- Weatherall DJ. The thalassemia syndromes. Tex Rep Biol Med. 1980; 40: 323–333.
- Vichinsky EP. Changing patterns of thalassemia worldwide. Ann N Y Acad Sci. 2005; 1054: 18–24.
- Weatherall DJ. Fortnightly review: The thalassaemias BMJ. 1997; 314: 1675.
- El-Harith E, Kuhanau W, Schimidtke J. Identification and clinical presentation of beta thalassemia mutations in the eastern region of Saudi Arabia. J Med Genet. 1999; 36: 936–937.
- Weatherall D. Phenotype-genotype relationships in monogenic disease: lessons from the thalassemias. Nat Rev Genet. 2001; 2: 245–255.
- Modell B. Update to epidemiology of hemoglobin disorders with special references to thalassemia. Thalassemia International Federation. Internet Transfus Med. 1995; 5: 247-258.
- Khan WA, Banu B, Amin SK. Prevalence of Beta thalassemia trait and Hb E trait in Bangladeshi school children and health burden of thalassemia in our population. Dhaku Shishu (Child). Hosp J. 2005; 21: 1–7.
- Abolghasemi H, Amid A, Zeinali S. Thalassemia in Iran: epidemiology, prevention, and management. J Pediatr Hematol Oncol 2007; 29: 233–238.
- Weathall DJ, Clegg JB. Inherited Haemoglobin Disorders: An increasing global problem. Bull World Health Organ. 2001; 79: 704-712.
- Shekhar HU, Kabir Y, Hossain MM. Blood transfusion mediated viral infections in thalassemic children in Bangladesh. J Med Sci. 2007; 7: 131-135.
- Mirmomen S, Alavian SM, Hajarizadeh B. Epidemiology of hepatitis B, hepatitis C, and human immunodeficiency virus infecions in patients with beta-thalassemia in Iran: a multicenter study. Arch Iran Med. 2006; 9: 319- 323.
- Benito A, Villegas A, Perez-Cano R, Bernal R. Beta thalassemia in southwestern Spain: high frequency of IVSI-1 mutation. Br J Haematol. 1996; 92: 336-338.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010: 12: 61–76.
- Black ML, Sinha S, Agarwal S, Colah R, Das R, Bellgard M, et al. A descriptive profile of beta-thalassaemia mutations in India, Pakistan and Sri Lanka. J Community Genet. 2010; 1: 149–157.
- Stephen Little. Amplification-Refractory Mutation System (ARMS) Analysis of Point Mutations. Hum Genet. 2001; 13: 8-9.
- Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, et al. Analysis of any point mutation in DNA. The Amplification Refractory Mutation System (ARMS). Nucl Acids Res. 1989; 17: 2503-2516.
- Weatherall DG, Clegg JB. The thalassaemia syndromes, 4th ed. Oxford: Blackwell Scientific Publication. 770–780.
- Hardison RC, Chui DH, Giardine B, Riemer C, Patrinos GP, Anagnou N, et al. Hb Var: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutatation. 2002; 19: 225–233.
- Chatterjee T, Chakravarty A, Chakravarty S, Mahmood Ahmed Chowdhury & Razia Sultana: Mutation Spectrum of β-Thalassemia and Other Hemoglobinopathies in Chittagong, Southeast Bangladesh: Hemoglobin. 2015; 39: 389–392.
- Black ML, Sinha S, Agarwal S, Colah R, Das R, Bellgard M, et al. A descriptive profile of beta-thalassaemia mutations in India, Pakistan and Sri Lanka. J Community Genet. 20.
- Waspi P, Winter WP. Geographic distribution of haemoglobin variants in South East Asia. Haemoglobin variants in human populations, Florida: CRC Press Inc. 1986; 2: 111-127.
- Galanello R, Eleftheriou A, Synodinos JT, Old J, Petrou M. Prevention of Thalassemias and other Haemoglobin disorders. Thalassemia International Federation Publications, Nicosia, Cyprus. 2003.
- Varawalla NY, Old GM, Venkateshanz SR, Weatherall DJ. The spectrum of beta thalassemia mutations on the Indian subcontinent- the basis of prenatal diagnosis. Brit J Hematol. 1991; 78: 242-247.
- Treisman T, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned b-thalassaemia genes. Nature. 1983; 302: 591–596.
- Kazazian HH, Orkin SH, Antonarakis SE. Molecular characterization of seven b-thalassemia mutations in Asian Indians. EMBO J. 1984; 3: 593–596.
- Cheng T, Orkin SH, Antonarakis SE. B-Thalassemia in Chinese: Use of in vivo RNA analysis and oligonucleotide hybridization in systematic characterization of molecular defects. Proc Natl Acad Sci USA, 1984; 81: 2821–2825.
- Kimura A, Matsunaga E, Takihara Y. Structural analysis of a b-thalassemia gene found in Taiwan. J Biol Chem. 1983; 258: 2748–2749.