
Letter to the Editor
J Blood Disord. 2014;1(4): 1019.
How to Find Gaucher Patients More Effectively - An Experience of One Haematooncology Department in Poland
Sokolowska B*
Hematooncology Department, Medical University of Lublin, Poland
*Corresponding author: Sokolowska B, Hematology Department, Medical University of Lublin, 20-081 Lublin, Staszica 11, Poland.
Received: December 26, 2014; Accepted: December 29, 2014; Published: December 31, 2014
Abstract
In this letter the author would like to share her experience concerning diagnostics of Gaucher Disease in Haematooncology Department of Medical University of Lublin in Poland.
Keywords: Gaucher disease; Diagnostics; DBS test
Abbreviations
GD: Gaucher Disease; DBS: Dried Blood Spot; GBA: Glucocerebrosidase; ERT: Enzyme Replacement Therapy; SRT: Substrate Reducing Therapy; MCV: Mean Corpuscular Volume; Hb: Hemoglobin; G/L: Giga/Liter; hr: hour; g: gram; mg: milligram; nmol: nanomol; pmol: picomol; dL: deciliter; fL: femtoliter
Letter to the Editor
Gaucher Disease (GD) is a progressive macrophage lipidosis caused by an autosomal recessive deficiency of lysosomal acid β glucosidase (glucocerebrosidase, GBA). Three major subtypes of GD have been described based on the absence (type 1) or presence (types 2 and 3) of neurological symptoms. The vast majorities of patients exhibit the non-neuronopathic, or type 1 form of the disease. Gaucher disease type 1 occurs in all ethnic groups, but it is the most common among people of Ashkenazi Jewish ancestry, with the prevalence of 1 in 450 compared to 1 in 40 000-60 000 in non-Jewish populations [1,2]. Enzyme replacement therapy was introduced in Poland in 1995. At that time this therapy was provided by the Institute of Pediatrics at Children's Health Center in Warsaw. Since 2009 the treatment of GD has been managed according to Therapeutic Health Program Treatment of Gaucher disease financed by The National Health Fund. A patient with newly diagnosed Gaucher disease is evaluated in hospital which is provided Therapeutic Program. Then all data concerning this patient is sent to Commission for Rare Disorders. This authority body approves the patient for the treatment. Finally the patient is treated in hospital in which he was previously evaluated. There are some guidelines regarding the treatment and evaluation of patients with GD. About 60 patients with GD are currently treated in Poland. It means that the number of GD patients is approximately 60. According to the prevalence of GD, estimated number of GD patients in our population should be 200. Conclusion: Gaucher disease is under diagnosed in our country. What is the reason for this situation?
In the era prior to the introduction of Enzyme Replacement Therapy (ERT) hematologists were generally physicians who treated patients with GD due to hematological symptoms like: thrombocytopenia, anemia, leukopenia and hepatosplenomagaly.
Nowadays hematologists remain at the forefront of specialists who patients with GD are referred to because of symptoms mentioned above.
However, most hematologists lack familiarity with recognition and management of GD mainly due to rare occurrence of the disease. It is believed that there is no chance of encountering such patients throughout an entire medical practice. Such beliefs contribute to diagnostic delays leading to severe complications that could have been prevented or reversed by appropriate therapy. We recorded the first GD patient in our center in 2003 [3]. A 43-years-old male was diagnosed because of moderate hepatosplenomegaly. The patient had complained of weakness and yellowish skin color for two years. Laboratory data revealed thrombocytopenia (platelet count 108G/L) and slightly elevated bilirubin level (1.68mg/dL). Gaucher cells were found in the bone marrow (Figures 1 & 2) as well as in the liver biopsy specimen.
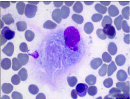
Figure 1: Gaucher cells in the bone marrow aspirates the patient mentioned above, Magnification x 100.
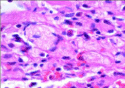
Figure 2: Gaucher cells in the bone marrow trephine biopsy done in the same patient, Magnification x 400.
The chitotriosidase serum level was greatly increased (11.5400 nmol/mg protein /hr, normal value <150, Lab.Warsaw). Glucocerebrosidase activity in leukocytes was within normal limits but when measured within cultured fibroblasts it showed deep deficiency. The patient's genotype was identified as N370S/c. ins.203insC. The patient started ERT in 2004.
The second patient, a 34-year-old female, was diagnosed in 2006, because of hypermenorrhea which had started five years earlier. Laboratory data revealed thrombocytopenia (platelet count 100G/l), normocytic anemia (Hb 11.2g/dL, MCV 90fL) and leukopenia (3.8- 4.0G/L). Ultrasound evaluation detected enlarged liver and spleen. Trephine biopsy of the bone marrow revealed diffuse infiltration with Gaucher cells. The chitotriosidase serum level was increased (6.110nmol/mg protein/hr). GBA activity in leukocytes was within normal limits. The assay measuring GBA activity in cultured fibroblasts confirmed the diagnosis of GD. The N370S/G377S genotype was identified. The patient started ERT in 2007.
The third patient had been under observation due to thrombocytopenia since childhood. When the patient was 36 years old a variant form of GD was diagnosed. This is a rare variant form caused by saposine C deficiency. The patient was treated with SRT (substrate reducing therapy) using miglustat for two years. The administration of miglustat failed to exert any favorable effects on the clinical condition, hematological parameters and glucosylceramide level in the serum. Thus the patient underwent splenectomy. The sister of the patient mentioned above was also diagnosed when she was 30 years old. She had the same rare variant of GD but she refused treatment. To intensify the diagnostics of GD we started to use DBS (dried blood spot) test [4]. From 16 June 2013 to 30 December 2014 twenty-eight patients underwent DBS test. All patients, except one, presented splenomegaly. Among these patients four demonstrated very low level of glucocerebrosidase (0, 90, 44, 65, normal value 200- 2000 pmol/spot*20hr, lab. Hamburg). Eight patients demonstrated only slightly lower than normal level of glucocerebrosidase (166- 186). These patients were presented to further diagnostics to genetic Counseling of Institute of Psychiatry and Neurology in Warsaw. Patients from the group mentioned above (with very low level of glucocerebrosidae) underwent another evaluation using DBS test. The first patient, a 29-year-old female was presented to hematologist because of thrombocytopenia (platelet count 60G/L) and splenomegaly. The activity of beta glucosidase was 0. Using DBS test we detected the following mutation: [703T>C]; [1226A>G], (p [Ser196Pro]; [Asn370Ser]. The patient started ERT on 10 October 2014. In two patients with GBA activity 90 and 44 respectively, molecular genetic testing revealed no pathogenic mutation. In the patient with GBA activity 65 in another DBS test normal enzyme activity was detected. The last three patients were presented to further diagnostics, especially to evaluate chitotriosidase activity.
Based on our center's experience mentioned above the following conclusions have been made: (1) GD is under diagnosed in Polish population, (2) Hematologists should expand their knowledge about GD because they remain the forefront of specialists whom patients with Gaucher disease are referred to. (3) Intensification of GD diagnostics, using simple test like DBS test, seems to be the most essential nowadays.
References
- Niederau C, Haussinger D. Gaucher's disease: a review for the internist and hepatologist. Hepatogastroenterology. 2000; 47: 984-997.
- Sokolowska B. Zaburzenia budowy monocytow i makrofagow. W: Antczak , Mysliwiec, Pruszczyk ,reds. Wielka Interna,Warszawa. 2011.
- Sokolowska B, Skomra D, Czartoryska B, Tomczak W, Tylki-Szymanska A, Gromek, et al. Gaucher disease diagnosed after bone marrow trephine biopsy - a report of two cases. Folia Histochem Cytobiol. 2011; 49: 352-356.
- Pompe Disease Diagnostic Working Group, Winchester B, Bali D, Bodamer OA, Caillaud C, Christensen E, et al. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008; 93: 275-281.