
Special Article: Coagulation Disorders
J Blood Disord. 2023; 10(1): 1071.
Fibrinolysis-Related Bleeding Troubles
Bashir BA*
Department of Hematology, Faculty of Medical Laboratory Sciences, Port Sudan Ahlia College, Sudan
*Corresponding author: Bashir Abdrhman Bashir Associate professor of Hematology, Chairman of Hematology Department Faculty of Medical Laboratory Sciences, Port Sudan Ahlia College, Port Sudan, Sudan
Received: December 14, 2022; Accepted: February 07, 2023; Published: February 14, 2023
Abstract
An intricate enzymatic process called fibrinolysis is used to eliminate blood clots and stop vascular clogging. Numerous cofactors that make up the fibrinolytic system regulate fibrin breakdown and uphold the hemostatic balance. Different disease events that promote prothrombotic or hemorrhagic states based on the form of aberration are linked to the dysregulation of fibrinolysis. This review is centered on fibrinolysis ailments that can be either heritable or acquired and affect both adults and children. A shortage of one of the fibrinolysis inhibitors deficiencies, such as Plasminogen Activator Inhibitor Type 1 (PAI-1) or 2-plasmin inhibitor, or an overabundance of one of the activators, such as tissue-type plasminogen activator or urokinase-type plasminogen activator, can lead in hyperfibrinolytic bleeding. Delayed bleeding following trauma, surgery and dental interventions is a hallmark of fibrinolytic illnesses with a bleeding phenotype. Bleeding in states like menorrhagia and epistaxis, which has significant fibrinolytic activity, is also frequent. The most extreme bleeding episodes are experienced by patients with 2-plasmin inhibitor deficiency. Interestingly, it was found that, particularly in patients with PAI-1 deficiency, hyperfibrinolytic diseases are linked to a high prevalence of obstetric problems such as miscarriage and preterm delivery. Due to ignorance and a lack of reliable diagnostic tools, hyperfibrinolytic diseases are likely to be underdiagnosed. The vast majority of individuals whose bleeding was deemed to be “of unknown origin” may have a hyperfibrinolytic disease. Because these conditions may typically be successfully treated with antifibrinolytic drugs, early detection is crucial.
Keywords: Fibrinolysis; Bleeding Disorders; 2-Plasmin Inhibitor; Plasminogen Activators Inhibitors -1; Hyperfibrinolysis
Introduction
Fibrinolysis is a sensitive and intricate enzymatic procedure designed to dissolve blood clots to localize and restrict clot development [1,2]. Fibrinolysis, which breaks down fibrin into soluble Fibrin Split Products (FSP), is regulated by serine proteases and regulatory protease inhibitors, which have the inverse impact on plasminogen conversion to plasmin, the active enzyme that fractures the fibrin clot [3]. Thus, the fibrinolytic framework is made up of both pro- and anti-fibrinolytic components, such as a2-plasmin inhibitor, Plasminogen Activator Inhibitor 1 (PAI-1), and Thrombin-Activatable Fibrinolysis Inhibitor (TAFI). Pro-fibrinolytic components include Tissue Plasminogen Activator (tPA) and Urokinase Plasminogen Activator (u-PA) [2]. The equilibrium between these two opposing enhancers is maintained by fibrinolysis under physiological statements; unusually limited or exaggerated fibrinolysis disturbs this equilibrium and can promote thrombosis or pathological hemorrhage, respectively [4]. Hypo- or hyper-fibrinolytic states can be genetic, more usually acquired, or induced by a single molecular flaw (Figure 1) [4,5]. In this overview, the main pathogenetic, laboratory, clinical, and features of hereditary and acquired hemorrhagic diseases linked to hyperfibrinolysis are outlined.
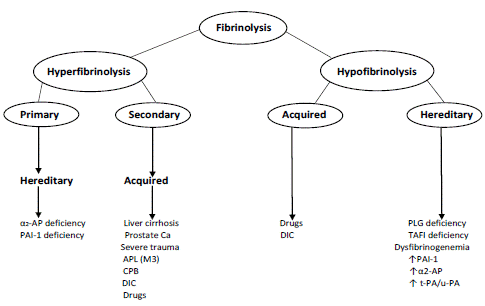
Figure 1: Overall survival, autologous stem cell transplant (ASCT) versus no ASCT (p=0.12).
Methodology
A thorough search for pertinent papers published before 30 November 2022 was conducted in EMBASE and PubMed. The methodology assessing the occurrences of clinical manifestations covered articles concerning at least one clinical indication of at least one patient with hemorrhagic disease of fibrinolysis. The references to the detected articles yielded more pertinent papers. The search method turned up 1377 articles. 1023 papers were determined to be eligible based on title and abstract after duplicates were removed. In (Figure 2), the study flow diagram is displayed. The present review covers the presence of clinical symptoms as well as details on the diagnosis and treatment of each ailment.
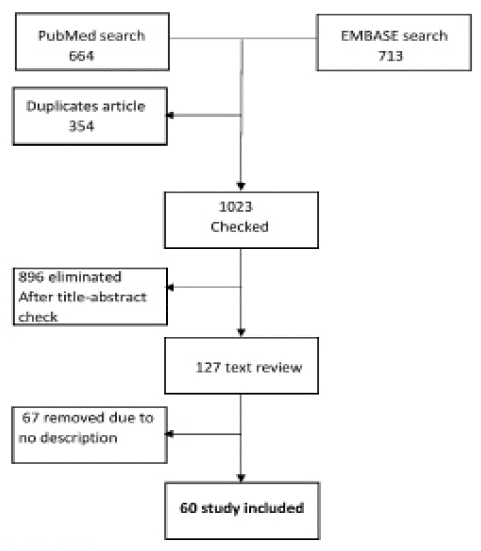
Figure 2: Study schedule.
Inherited Bleeding Episodes of Fibrinolysis Framework
a2-plasmin inhibitor(a2-antiplasmin) deficiency
Background: The fundamental biological inhibitor of plasmin is the 2-plasmin inhibitor, a single-chain glycoprotein of 70 kDa manufactured by the liver [6]. Genetical 2-antiplasmindeficiency is an uncommon autosomal recessive syndrome (chromosome17) hallmarked by pathological bleeding brought on by premature disintegration of hemostatic plugs. Usually manifests as rebleeding after trauma or medical interventions [7,8]. By attaching to the lysine-binding site of plasminogen and fully blocking plasminogen's ability to bind to fibrin, the 2-plasmin inhibitor completely suppresses fibrinolysis. It also covalently binds to fibrin via activated factor XIII (FXIIIa), which stops plasmin from breaking down fibrin [1,9]. Because coagulation biomarkers are normal yet the euglobulin clot lysis time is quick due to unrestrained plasmin activity, it is challenging to diagnose 2-plasmin inhibitor deficiency. The diagnosis is supported by the functional and immunologic plasma experiments of the inhibitor [8]. Two distinct types of 2-antiplasmin deficiency are caused by various mutations in the gene encoding the protein: a quantitative defect (type I), evidenced by a similar decline in plasma levels of antigen and activity, and a qualitative defect (type II), owing to the lower functional activity in contrast with type I [8].
Clinical manifestation: Symptoms of bleeding, such as prolonged bleeding from wounds, epistaxis, and gingival bleeding, are described in case reports of Sudanese infants with a congenital deficit of a2-plasmin inhibitor [9]. Another vast spectrum of bleeding manifestations, which include umbilical cord bleeding, prolonged wound bleeding, epistaxis, gingival bleeding, subcutaneous and intramuscular hematomas, hematuria, hemarthroses, and central nervous system bleeding, are described in case reports of both children and adults with inherited deficiency of 2-plasmin inhibitor [10-14]. However, only homozygotes have a significant tendency to bleed, which is typically severe and begins in childhood, whereas hemorrhagic events are reported to happen only following trauma, dental extractions, or surgery in heterozygotes [8].
Diagnosis and management: A lack of knowledge on 2-plasmin inhibitor deficiency may cause the diagnosis to be underestimated. The absence of aberrations in screening assays may make this worse. Both carriers and homozygous individuals can have typical euglobulin clot lysis times, albeit this is uncommon [15,16]. Owing to the lack of concentration of 2-plasmin inhibitor in the euglobulin fraction following acidification of plasma, this low sensitivity for 2-plasmin inhibitor deficit may exist [17]. Directed 2-plasmin inhibitor confirmation assays should be run when a deficiency is suspected based on a shorter clot lysis time and/or suspicious background. A deficit may be qualitative (type II), resulting in low activity levels with normal or barely lowered antigen concentrations, or quantitative (type I), resulting in a concurrent drop in 2-plasmin inhibitor activity and antigen [10]. The antifibrinolytic medicine Tranexamic Acid (TXA) is useful for treating acute bleeding episodes as well as stopping bleeding during or after invasive or surgical operations. When an urgent boost in this fibrinolysis inhibitor is required, Fresh-Frozen Plasma (FFP) may be administered as an option or a supplement to antifibrinolytic medication [18]. Since plasma that has been treated with a solvent detergent has lower levels of 2-antiplasmin, it is not advisable to use it to cure 2-antiplasmin shortage [5]. Presently, omniplasma is replacing FFP in many nations; however, omniplasma has lower a2-plasmin inhibitor levels and quicker Rotational Thromboelastometry (ROTEM) lysis durations that have been comparable to FFP, with a drop of more than 50%. As a result, it is not advisable to utilize omniplasma to treat a2-plasmin inhibitor deficiency [19]. Desmopressin acetate ought to be averted since it can cause the release of plasminogen activator [20].
Plasminogen Activator Inhibitor-1 Deficiency
Background: PAI-1 Is a single-chain glycoprotein of 52 kDa generated in the liver (chromosome 7) that limits plasmin proteolytic activity by frustrating the plasminogen activators tPA and uPA [21-23]. Genetical PAI-1 deficiency is an autosomal recessive trait that can manifest as either a qualitative (dysfunctional PAI-1 synthesis with detectable protein but reduced or absent functional activity) or a quantitative (reduced or missing protein production) characteristic [22]. Even though PAI-1's primary function is to suppress fibrinolysis, it also serves other roles in inflammation as an acute-phase protein. Excessive PAI-1 levels, in contrast to low PAI-1 levels, are linked to atherosclerosis and coronary artery disease, metabolic syndrome, fibrosis, and poor outcomes for many cancer types [24]. Antigen and activity levels should be examined when assessing a patient for PAI-1 deficiency because the deficiency might be either qualitative or quantitative [25]. Genetic PAI-1 deficiency is difficult to diagnose because screening coagulation tests are usually normal [2].
Clinical manifestations: Rarely do these patients experience spontaneous bleeding; instead, abnormal bleeding only occurs after trauma or surgery. Congenital PAI-1 deficiency has also been linked to a predisposition to bleed in infants [22] and kids [26]. While heterozygotes typically do not exhibit bleeding symptoms, homozygotes with PAI-1 deficiency appear to have mild to moderate bleeding conditions [27-29]. In females with PAI-1 deficiency, menorrhagia and obstetric problems (miscarriages and premature deliveries) have been documented [30]. Women with a PAI-1 deficit seem to have obstetric problems more frequently. Even though animal studies have indicated that PAI-1 and the associated proteolytic mechanism have a role in follicular wall breakdown during ovulation, as well as in fertilization, embryo implantation, embryogenesis, and angiogenesis [31,32], the exact cause of this remains unclear. In consequence, heterozygous PAI-1 deficiency is typically asymptomatic, however homozygous PAI-1 deficiency is linked to severe bleeding issues and maternal difficulties.
Diagnosis and management: The exact diagnosis of a PAI-1 deficiency is sometimes delayed due to common screening tests and a poorer understanding of the condition. The assays for PAI-1 activity that are now available lack discrimination in the lowest range since their normal limit is zero [22]. The diagnosis is based on the outcomes of antigenic (enzyme-linked immunosorbent assay, ELISA) and functional (chromogenic test) PAI-1 assays, even though the euglobulin clot lysis time is brief and whole blood clotting tests like the thromboelastogram are atypical [22]. Also, a precise diagnosis may become more challenging due to PAI-1's diurnal oscillation, which has maximum levels in the morning and lowest levels in the afternoon [30]. TXA is the cornerstone medication for the treatment of acute bleeding. This medication, especially when used prophylactically, is very successful at reducing bleeding in patients having surgery, especially oral and urogenital operations. Hormonal therapy, as well as long-term preventative antifibrinolytic medication, if indicated, can successfully treat persistent menorrhagia. TXA is typically administered at doses of 25 mg/kg body mass every 8 hours. The typical length of treatment for preventing and treating bleeds is 3-4 days for small procedures and 5-7 days for large surgeries with bleeds. It is best to avoid desmopressin acetate since it can cause plasminogen activators to be secreted from endothelial cells [5].
Quebec Platelet Disorder
Background: The Quebec Platelet Syndrome (QPS) or (Urokinase-type plasminogen activator excess) is a rare autosomal dominant bleeding disorder that is accompanied by a mildly diminished platelet count. It is brought on by the upregulation of uPA and increased storage of the protein in platelet granules, which results in increased release of uPA and hyperfibrinolysis [33-35]. On chromosome 10 is where the hypothetical gene PLAU (urokinase plasminogen activator) is located. Excessive uPA expression and storage in platelets are the root causes of hyperfibrinolysis [36]. When megakaryocyte differentiation occurs, the genetic flaw increases the transcription of the PLAU allele on chromosome 10 [37]. Although bleeding was once believed to be triggered by a qualitative platelet factor V deficiency, the first case was first documented in 1984. As a result, factor V Quebec was the first name given to the condition [37]. Unknown to the globe is how common the condition is.
Clinical manifestations: Clinically, Quebec platelet syndrome patients often exhibit delayed bleeding after trauma or surgical and dental treatments, but they can also exhibit easy bruising, epistaxis, hematuria, menorrhagia, and joint bleeds. Even with treatment, bleeding can occasionally be excessive and uncontrolled [38].
Diagnosis and management: Owing to the great penetrance of this autosomal dominant condition, the family background will typically be favorable and arouse suspicion in cases of bleeding. Excessive platelet uPA content, which is released following platelet activation, is a hallmark of the condition. uPA levels in plasma are normal or just mildly elevated during rest [35]. Unknown slight thrombocytopenia can coexist with the condition. Coagulation diagnostic tests are often normal, though factor (F) V deficiency can occasionally occur. FV of platelets is lowered [37]. Functional platelet tests frequently show a distinctive pattern of absent epinephrine-induced platelet aggregation and diminished platelet aggregation in the presence of ADP and collagen. This trend is likely caused by the overabundance of uPA, which causes plasminogen to be activated in platelets. Then, plasmin can destroy fibrinogen, FV, FVIII, and von Willebrand factor [35]. When the Urinary PA is not elevated, the whole-blood clot lysis time is normal [34,39]. It is now advisable to use a PCR assay for the QPD mutation when a diagnosis is anticipated because the genetic abnormality has been uncovered and other diagnostics are unpredictable and challenging [40]. Antifibrinolytic pharmaceuticals, the cornerstone of both hemorrhage prevention and treatment, should be resisted in hematuric patients due to the possibility of renal occlusion from urinary tract clot formation [34]. Using fibrinolytic antagonists as a preventative measure has been helpful during labor as well [38]. Although platelet transfusions have been utilized, they might not be helpful when there is significant bleeding [35].
Tissue-Type Plasminogen Activator Excess
When tPA antigen levels are normal, patients with a congenital PAI- 1 deficiency may have elevated amounts of tPA activity [41,42]. These patients experienced profuse bleeding following trauma and surgery, as well as easy bruising or spontaneous he matomas in some circumstances. In the absence of any bleeding symptoms, family members of a patient with an overabundance of tPA had high tPA levels and a reduced euglobulin clot lysis time [30]. We advocate using an activity assay for diagnosis versus an antigen assay because currently, available antigen assays evaluate both the free form and complexes between tPA and PAI-1 in addition to the free form.
Acquired Bleeding Episodes of Fibrinolysis Framework
Liver cirrhosis
Background: Major clinical bleeding is a frequent complication of severe liver disease owing to a variety of factors, such as endothelial dysfunction and thrombocytopenia brought on by splenic sequestration, portal hypertension with varices developing, and a decreased synthesis of coagulation factors and their inhibitors brought on by impaired hepatic function [43–55]. As many as 50% of patients with end-stage liver disease may [46-48] experience a hyperfibrinolytic condition, which is a frequent finding in liver cirrhosis and is linked to mucocutaneous and gastrointestinal bleeding [49]. The enhanced endothelial release and decreased hepatic clearance of tPA, as well as the decreased production of TAFI, a2-plasmin inhibitor, and PAI-1, are clinically significant factors that strongly stimulate the fibrinolytic system [50-53].
Diagnosis and management: Elevated tPA and D-dimer levels constituted the only indicators of bleeding in individuals with hepatic cirrhosis and esophageal varices but no upper gastrointestinal bleeding was monitored [30]. The effective use of antifibrinolytic medications in cirrhotic patients, particularly those with mucosal or gastrointestinal bleeding, provides additional evidence for the therapeutic significance of primary hyperfibrinolysis [54]. Patients getting orthotopic liver transplantation who exhibit accelerated fibrinolysis (particularly during the anhepatic phase) together with elevated tPA and decreased plasma levels of a2-plasmin inhibitor may also be at risk for serious bleeding instances due to hyperfibrinolysis [30]. Antifibrinolytic medication helps to decrease blood loss and the requirement for perioperative transfusions in this type of surgery, according to a Cochrane review [55].
Trauma
Background: Trauma contributes to upwards of 6 million fatalities globally each year [56], making it a major global health burden. Interestingly, within the first several hours following trauma, bleeding accounts for close to 50% of fatalities from trauma [56]. Up to 25% of critically injured patients who report to the emergency room have a hemostatic problem, which is a typical indication in trauma patients [56-59]. This early extreme traumatic coagulopathy is an intrinsic event that is primarily characterized by primary hyperfibrinolysis, fibrinogen depletion, augmented thrombin production, platelet dysfunction, and endothelial hypoperfusion and tissue injury [60–62]. A prominent role of rapid fibrinolysis in the pathogenesis of acute traumatic coagulopathy is supported by several lines of evidence [4]. When hyperfibrinolysis was identified by viscoelastic testing upon arrival to the emergency room, Schochl and colleagues [63] found a mortality rate of almost 88% in trauma patients.
Diagnosis and management: In the bulk of trauma victims, primary hyperfibrinolysis was detected by thromboelastometry and other investigations (plasmin-antiplasmin complex and D-dimer levels), according to research by Raza and colleagues [64]. Although the direct fibrinogenolysis that results from the disturbance of fibrinogen metabolism brought on by hypothermia and metabolic acidosis have been suggested as plausible explanation, the specific mechanism underpinning fibrinolysis activation in severe trauma is still not well understood [59]. Additionally, it appears that the activation of the protein C pathway is crucial in promoting systemic fibrinolysis and causing fibrinogen deficiency [56]. The large production of thrombin and the formation of its complex with thrombomodulin on the endothelium surface after trauma significantly increase protein C activation [63]. Iatrogenic factors also can potentially contribute to hyperfibrinolysis. Rapid thrombin production, high amounts of circulating adrenaline, bradykinin, vasopressin, and cardiopulmonary bypass all contribute to a state of hyperfibrinolysis [65]. Several studies [66–68] looked at the therapeutic potential of antifibrinolytic drugs, particularly TXA, to counteract hyperfibrinolysis in patients with severe trauma. According to the Cochrane review on antifibrinolytic medications for acute traumatic injury, TXA limited the risk of death by 10% (RR 0.90, 95% CI 0.85-0.96; p = 0.002) without raising the risk of side effects in the assessment of various trials involving 20,548 patients [67]. TXA has also undergone a thorough evaluation in cases of traumatic brain injury. In patients with mild to moderate head injuries; TXA reduced the incidence of head-injury-related mortality (RR 0.78, 95% CI 0.64-0.95). Similar to CRASH-2, individuals with minor and moderate head damage responded better to medical intervention than delayed treatment (p = 0.005), with a 10% reduction in response for every 20 minutes of delay [67].
Acute Promyelocytic Leukemia (APL)
Background: Acute Promyelocytic Leukemia (APL) is a unique subtype of acute myelogenous leukemia due to its unusual molecular, morphological, and clinical presentations [69]. A translocation involving chromosomes 15 and 17 that enables the fusion of the Promyelocytic Leukemia protein gene (PML) with the Retinoic Acid Receptor (RAR) gene has been recognized as the sole genetic abnormality causing APL. APL is caused by the expressed PML-RAR-fusion protein, which prevents myeloid precursor cells from differentiating and extends their survival [54]. An increased frequency of serious bleeding problems is a characteristic of APL [69]. The molecular mechanism behind the hemostatic abnormalities in APL, which include elevated circulating levels of uPA and tPA and lower levels of PAI-1, a2-plasmin inhibitor, and TAFI, have been clarified in the last ten years [70-76]. In turn, the S100 protein, a calcium-binding protein belonging to the S100 family, forms a heterotetrameric complex with annexin A2, a protein receptor with a great affinity for plasminogen and tPA and a powerful cofactor for the conversion of plasminogen to plasmin. The expression of the S100 protein is boosted by the fusion protein PML-RAR. As a result, the S100-annexin A2 complex that is attached to the surface helps to activate plasminogen while also preventing plasmin from being inhibited [77]. Remarkably, PML-RAR- can also significantly boost annexin A2 expression [78]. Clinically, high levels of tissue factor and cancer procoagulant expressed by leukemic promyelocytes lead to activation of coagulation, which in turn causes enhanced fibrinolysis and decreased clotting factors [75]. This activation of coagulation is what causes the severe hemorrhagic diathesis seen in APL patients.
Diagnosis and management: The characteristic laboratory evidence of APL was reflected by a substantial drop in fibrinogen levels along with high FDPs as a result of this two-fold illness mechanism. All-Trans Retinoic Acid (ATRA) was launched for the cure of APL, significantly reducing early hemorrhagic mortality in newly diagnosed patients [71]. ATRA can decrease S100 and annexin A2 overexpression by encouraging the differentiation of leukemic blasts, which helps to rectify the hemostatic defect by restricting plasmin production on the surfaces of APL cells [72]. Antifibrinolytic medicines should have a positive impact given the prominent role of hyperfibrinolysis in the etiology of bleeding in APL [73], but the available information is few and somewhat contradictory [80]. The most retrospective trial of antifibrinolytics in APL found no improvement in transfusion requirements [81]. Nevertheless, tiny case series on their usage in APL have shown some improvement in laboratory indices of fibrinolysis and a diminution in the need for transfusions [81]. Moreover, certain investigations have highlighted some questions about the safety of these medications in APL, especially concerning thrombotic risk [82].
Post-Partum Hemorrhage
Background: A model definition of post-partum hemorrhage, a leading cause of maternal death globally, is blood loss of 500 mL or more within 24 hours. This condition is distinguished by the beginning of hyperfibrinolysis early on [83,84]. Enhanced endothelial t-PA synthesis and PAI-1 inhibition by protein C activation appear to be comparable to the pathophysiology of increased fibrinolytic activity documented in severe trauma [85]. FPD levels rise in the hours after delivery in healthy pregnancies, but this increment is significantly greater in women who have post-partum hemorrhage [84].
Diagnosis and management: The recent discovery of elevated D-dimer levels and plasmin-antiplasmin complexes in the early post-partum period [86] further supports the substantial role provided by fibrinolysis in post-partum bleeding. With this context, it makes clinical sense to employ TXA, an antifibrinolytic drug, to treat post-partum hemorrhage [87]. In the randomized, placebo-controlled trial (WOMAN, World Maternal Antifibrinolytic), 20,060 post-partum hemorrhaging women who had undergone vaginal or cesarean delivery were randomly assigned to receive either TXA (1 g TXA intravenously as quickly as possible, followed by a further 1 g of TXA if bleeding persisted after 30 min or relaunched within 24 h of the initial dose) or a placebo [88]. TXA reduced bleeding-related mortality without having any negative side effects (RR 0.81, 95% CI 0.65-1.00; P= 0.045), especially when administered as soon as feasible after the commencement of bleeding. Early TXA administration may have a favorable impact in postpartum hemorrhage as well as in the trauma configuration because it prevents residual fibrinogen stores (already depleted by the underlying bleeding condition) from further decreasing due to hyperfibrinolysis, preserving their ability to form a stable clot [4].
Unknown-Cause Bleeding with Fibrinolysis
Even after doing all available screening and conformational testing, a sizable number of people with a tendency to bleed and a family history of bleeding issues remain undiagnosed. There may be a subset of people with a hyperfibrinolytic condition in this group. Additionally, numerous investigations have shown that people with a tendency to bleed for unclear reasons have a different fibrinolytic balance. Fewer preoperative levels of PAI-1 and reduced tPA-PAI-1 levels were correlated to postoperative blood loss in a group of patients undergoing cardiac surgery [89]. On the other token, there were no variations in PAI-1 levels between patients and healthy controls in research examining fibrinolysis in patients with a mild-to-moderate bleeding propensity of unclear cause. Additionally, individuals with a predisposition to bleed have low levels of PAI-1 activity, which is prevalent in the general population, and these levels are not vastly different from those of blood donors and healthy controls [90].
Conclusion
The occurrence of hyperfibrinolytic syndromes as a cause of bleeding recorded in the literature is minimal, but the propensity to bleed is typically defined by a trend of delayed bleeding after trauma or surgery and mucocutaneous bleeding, such as menorrhagia and epistaxis. Nonetheless, as they often also present with mucocutaneous bleeding, it can be challenging to clinically distinguish them from other bleeding troubles, such as von Willebrand disease or platelet disorders. If fibrinolytic bleeding consequences are not appropriately managed with antifibrinolytic medication, they can be quite serious and increase morbidity and death.
Patients with a surplus of boosters of fibrinolysis and victims with a lack of natural physiological inhibitors can be classified as having the same type of fibrinolytic disease. a2-plasmin inhibitor deficiency is responsible for the most extreme clinical phenotype. Interestingly, obstetric problems are associated with PAI-1 deficiency due to an unidentified underlying mechanism. Obstetric troubles are not yet known to arise about the other fibrinolytic illnesses, making this an intriguing area for further study.
The occurrence of fibrinolytic disorders is likely larger than previously thought due to the lack of identification of these conditions and the challenges in making the proper diagnosis. Because there isn't a sensitive, reliable fibrinolysis test, proper diagnosis is greatly hampered. This explains both the PAI-1 activity and antigen levels as well as the euglobulin clot lysis time.
A hyperfibrinolytic disease cannot be ruled out by the normal findings of these tests. Additionally, these current fibrinolytic techniques are only possible in specialist laboratories. Universal assays may be able to solve this issue because they are typically more sensitive and may evaluate multiple routes in a single test. These assays' limited availability is a drawback. Additionally, next-generation sequencing methods of genes involving the entire coagulation spectrum may help to clarify suspected fibrinolytic diseases. Patients with hemorrhage of unclear origin who undergo fibrinolytic assays get conflicting results. Overall, further research in this area would be helpful because an imbalance in the fibrinolytic system may account for a predisposition to bleed more frequently than is currently thought.
Ultimately, the inability to address bleeding issues with focused therapy without a precise and correct diagnosis results in the needless use of unnecessary blood products and pricey factor concentrates. Since they are infrequent and laboratory diagnostics are poor, disorders of fibrinolysis shouldn't be regularly screened for in patients with a propensity to bleed. Whenever there is a considerable index of suspicion, fibrinolysis examinations ought to be carried out in a specialized lab.
References
- Lin H, Xu L, Yu S, Hong W, Huang M, et al. Therapeutics targeting the fibrinolytic system. Exp Mol Med. 2020; 52: 367–379.
- Franchini M, Zaffanello M, Mannucci PM. Bleeding Disorders in Primary Fibrinolysis. Int J Mol Sci. 2021; 22: 7027.
- Longstaff C, Kolev K. Basic mechanisms and regulation of fibrinolysis. J Thromb Haemost. 2015; 13: S98–S105.
- Franchini M, Mannucci PM. Primary hyperfibrinolysis: Facts and fancies. Thromb Res. 2018; 166: 71–75.
- Jain S, Acharya SS. Inherited disorders of the fibrinolytic pathway. Transfus Apher Sci. 2019; 58: 572–577.
- Carpenter SL, Mathew P. Alpha2-antiplasmin and its deficiency: Fibrinolysis out of balance. Haemophilia. 2008; 14: 1250–4.
- Koie K, Kamiya T, Ogata K, Takamatsu J. Alpha2-plasmin-inhibitor deficiency (Miyasato disease). Lancet. 1978; 2: 1334–6.
- Welch SK, Francke U. Assignment of the human alpha 2-plasmin inhibitor gene (PLI) to chromosome 17, region pter-p12, by PCR analysis of somatic cell hybrids. Genomics. 1992; 13: 213–4.
- Favier R, Aoki N, de Moerloose P. Congenital alpha(2)-plasmin inhibitor deficiencies: A review. Br J Haematol. 2001; 114: 4–10.
- Yoshioka A, Kamitsuji H, Takase T, Iida Y, Tsukada S, et al. Congenital Deficiency of _2-Plasmin Inhibitor in Three Sisters. Pathophysiol Haemost Thromb. 1982; 11: 176–184.
- Kordich L, Feldman L, Porterie P, Lago O. Severe hemorrhagic tendency inheterozygous a2-antiplasmin deficiency. Thromb Res. 1985; 40: 645–651.
- Kluft C, Vellenga E, Brommer EJ, Wijngaards G. A familial hemorrhagic diathesis in a Dutch family: An inherited deficiencyof alpha 2-antiplasmin. Blood. 1982; 59: 1169–1180.
- Maino A, Garagiola I, Artoni A, Al-Humood S, Peyvandi F. A novel mutation of a2plasmin inhibitor gene causes aninherited deficiency and a bleeding tendency. Haemophilia. 2007; 14: 166.
- Matrane W, Bencharef H, Oukkache B. Congenital Alpha-2 Antiplasmin Deficiency: A Literature Survey and Analysis of 123 Cases. Clin Lab. 2020; 66.
- Griffin GC, Mammen EF, Sokol RJ, Perrotta AL, StoyanovichA, Abildgaard CF. Alpha 2-antiplasmin deficiency. An overlookedcause of hemorrhage. Am J Pediatr Hematol Oncol. 1993; 15: 328–30.
- Hanss MM, Farcis M, Ffrench PO, de Mazancourt P, Dechavanne M. A splicing donor site point mutation in intron 6 ofthe plasmin inhibitor (alpha2 antiplasmin) gene with heterozygous deficiency and a bleeding tendency. Blood Coagul Fibrinolysis. 2003; 14: 107–11.
- Smith AA, Jacobson LJ, Miller BI, Hathaway WE, Manco-Johnson MJ. A new euglobulin clot lysis assay for global fibrinolysis.Thromb Res. 2003; 112: 329–37.
- Morimoto Y, Yoshioka A, Imai Y, Takahashi Y, Minowa H, et al. Haemostatic management of intraoral bleeding in patients with congenital deficiency of alpha2-plasmin inhibitor or plasminogen activator inhibitor-1. Haemophilia. 2004; 10: 669–674.
- van Beers JJ, van Egmond LT, Wetzels RJ, Verhezen PW, Beckers EA, et al. Increased coagulation and fibrinolytic potential of solventdetergent plasma: a comparative study between Omniplasma and fresh frozen plasma. Vox Sang. 2016; 111: 33–42.
- Shahian DM, Levine JD. Open-heart surgery in a patient with heterozygous alpha 2-antiplasmin deficiency. Perioperative strategies in the first reported case. Chest. 1990; 97: 1488–90.
- Rijken DC. Plasminogen activators and plasminogen activator inhibitors: Biochemical aspects. Baillière’s Clin Haematol. 1995; 8: 291–312.
- Mehta R, Shapiro AD. Plasminogen activator inhibitor type 1 deficiency. Haemophilia. 2008; 14: 1255–1260.
- Urano T, Suzuki Y, Iwaki T, Sano H, Honkura N, et al. Recognition of Plasminogen Activator Inhibitor Type 1 asthe Primary Regulator of Fibrinolysis. Curr Drug Targets 2019; 20(16): 1695–1701.
- Iwaki T, Urano T, Umemura K. PAI-1, progress in understanding the clinical problem and its aetiology. Br J Haematol. 2012; 157: 291–8.
- Dieval J, Nguyen G, Gross S, Delobel J, Kruithof EK. A lifelong bleeding disorder associated with a deficiency of plasminogen activator inhibitor type 1. Blood. 1991; 77: 528–32.
- Minowa H, Takahashi Y, Tanaka T, Naganuma K, Ida S, et al. Four cases of bleeding diathesis in children due to congenital plasminogen activator inhibitor-1 deficiency. Haemostatis. 1999; 29: 286–291.
- Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: Characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997; 90: 204–208.
- Lee MH, Vosburgh E, Anderson K, McDonagh J. Deficiency of plasma plasminogen activator inhibitor 1 results in hyperfibrinolytic bleeding. Blood. 1993; 81: 2357–2362.
- Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Brief report: Complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. N Engl J Med. 1992; 327: 1729–1733.
- Saes JL, Schols SEM, Van Heerde WL, Nijziel MR. Hemorrhagic disorders of fibrinolysis: A clinical review. J Thromb Haemost. 2018; 16: 1498–1509.
- Heiman M, Gupta S, Shapiro AD. The obstetric, gynaecological and fertility implications of homozygous PAI-1 deficiency: single-centre experience. Haemophilia. 2014; 20: 407–12.
- Bajou K, Herkenne S, Thijssen VL, D’Amico S, Nguyen NQ, et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat Med. 2014; 20: 741–7.
- Blavignac J, Bunimov N, Rivard GE, Hayward CP. Quebec Platelet Disorder: Update on Pathogenesis, Diagnosis, and Treatment. Semin Thromb Hemost. 2011; 37: 713–720.
- Diamandis M, Veljkovic DK, Maurer-Spurej E, Rivard GE, Hayward CP. Quebec platelet disorder: Features, pathogenesisand treatment. Blood Coagul Fibrinolysis. 2008; 19: 109–119.
- Hayward CP, Rivard GE. Quebec platelet disorder. Expert Rev Hematol. 2011; 4: 137–141.
- Diamandis M, Paterson AD, Rommens JM, Veljkovic DK, Blavignac J, et al. Quebec platelet disorder is linked to the urokinase plasminogen activator gene (PLAU) and increases expression of the linked allele in megakaryocytes. Blood. 2009; 113: 1543–1546.
- Kahr WH, Zheng S, Sheth PM, Pai M, Cowie A, et al. Platelets from patientswith the Quebec platelet disorder contain and secrete abnormal amounts of urokinase-type plasminogen activator. Blood. 2001; 98: 257–65.
- McKay H, Derome F, Haq MA, Whittaker S, Arnold E, et al. Bleeding risks associated with inheritance of the Quebec platelet disorder. Blood. 2004; 104: 159–165.
- Diamandis M, Veljkovic DK, Derome F, Rivard GE, Hayward CPM. Evaluation of urokinase plasminogen activator in urinefrom individuals with Quebec platelet disorder. Blood Coag Fibrinol. 2008; 19: 463–4.
- Paterson AD, Rommens JM, Bharaj B, Blavignac J, Wong I, et al. Persons with Quebec platelet disorder have a tandem duplication of PLAU, the urokinase plasminogen activator gene. Blood. 2010; 115: 1264–6.
- Matsui H, Takahashi Y, Matsunaga T, Tanaka-Horie T, Minowa H, et al. Successful arthroscopic treatment of pigmented villonodularsynovitis of the knee in a patient with congenital deficiency ofplasminogen activator inhibitor-1 and recurrent haemarthrosis. Haemostasis. 2001; 31: 106–12.
- Zhang ZY, Wang ZY, Dong NZ, Bai X, Zhang W, Ruan CG.A case of deficiency of plasma plasminogen activator inhibitor-1 related to Ala15Thr mutation in its signal peptide. Blood Coagul Fibrinolysis. 2005; 16: 79–84.
- Tripodi A, Primignani M, Mannucci PM, Caldwell SH. Changing Concepts of Cirrhotic Coagulopathy. Am J Gastroenterol. 2017; 112: 274–281.
- Roberts LN, Patel RK, Arya R. Haemostasis and thrombosis in liver disease. Br J Haematol. 2010; 148: 507–521.
- Davis JP, Caldwell SH, Intagliata NM. Coagulation Pathways, Hemostasis, and Thrombosis in Liver Failure. Semin Respir Crit Care Med. 2018; 39: 598–608.
- Tripodi A, Fracanzani AL, Primignani M, Chantarangkul V, Clerici M, et al. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J Hepatol. 2014; 61: 148–154.
- Mannucci PM, Tripodi A. Hemostatic defects in liver and renal dysfunction. Hematology. 2012; 2012: 168–173.
- Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011; 365: 147–156.
- Leebeek FW, Rijken DC. The fibrinolytic status in liver diseases. Semin Thromb Hemost. 2015; 41: 474–480.
- Ferro D, Violi F. Clotting Activation and Hyperfibrinolysis in Cirrhosis: Implication for Bleeding and Thrombosis. Semin Thromb Hemost. 2013; 39: 426–433.
- Bennani-Baiti N, Daw HA. Primary hyperfibrinolysis in liver disease: A critical review. Clin Adv Hematol Oncol. 2011; 9: 250–252.
- von Meijenfeldt FA, Lisman T. Fibrinolysis in Patients with Liver Disease. Semin Thromb Hemost. 2021; 47: 601-609.
- Tripodi A. Hemostasis in acute and chronic liver disease. Semin Liver Dis. 2017; 37: 28–32.
- Breen KA, Grimwade D, Hunt BJ. The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol. 2011; 156: 24–36.
- Henry DA, Carless PA, Moxey AJ, O’Connell D, Stokes BJ, et al. Anti-fibrinolytic use for minimizing perioperative allogeneic blood transfusion. Cochrane Database Syst Rev. 2011.
- Gall LS, Brohi K, Davenport RA. Diagnosis and Treatment of Hyperfibrinolysis in Trauma (A European Perspective). Semin Thromb Hemost. 2017; 43: 224–234.
- Maegele M, Schöchl H, Cohen MJ. An update on the coagulopathy of trauma. Shock. 2014; 41: 21-5.
- Madurska MJ, Sachse KA, Jansen JO, Rasmussen T, Morrison JJ. Fibrinolysis in trauma: A review. Eur J Trauma Emerg Surg. 2018; 44: 35–44.
- Gall LS, Davenport RA. Fibrinolysis and antifibrinolytic treatment in the trauma patient. Curr Opin Anaesthesiol. 2018; 31: 227–233.
- Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, et al. Primary Fibrinolysis Is Integral in the Pathogenesis of the Acute Coagulopathy of Trauma. Ann Surg. 2010; 252: 434–444.
- Giordano S, Spiezia L, Campello E, Simioni P. The current understanding of trauma-induced coagulopathy (TIC): A focusedreview on pathophysiology. Intern Emerg Med. 2017; 12: 981–991.
- Davenport RA, Brohi K. Cause of trauma-induced coagulopathy. Curr Opin Anaesthesiol. 2016; 29: 212–2199.
- Schöchl H, Frietsch T, Pavelka M, Jámbor C. Hyperfibrinolysis After Major Trauma: Differential Diagnosis of Lysis Patterns and Prognostic Value of Thrombelastometry. J Trauma Inj Infect Crit Care. 2009; 67: 125–131.
- Raza I, Davenport R, Rourke C, Platton S, Manson J, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013; 11: 307–314.
- Yavari M, Becker RC. Coagulation and fibrinolytic protein kinetics in cardiopulmonary bypass. J Thromb Thrombolysis. 2009; 27: 95–104.
- Ramirez R, Spinella PC, Bochicchio GV. Tranexamic Acid Update in Trauma. Crit Care Clin. 2017; 33: 85–99.
- Ker K, Roberts I, Shakur H, Coats TJ. Antifibrinolytic drugs for acute traumatic injury. Cochrane. 2015; 5: CD004896.
- Franchini M, Mannucci PM. The never-ending success story of tranexamic acid in acquired bleeding. Haematologica. 2020; 105: 1201–1205.
- Kwaan HC. The unique hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost. 2014; 40: 332–336.
- Choudhry A, DeLoughery TG. Bleeding and thrombosis in acute promyelocytic leukemia. Am J Hematol. 2012; 87: 596–603.
- Kwaan HC, Cull EH. The coagulopathy in acute promyelocytic leukaemia—What have we learned in the past twenty years. Best Pr Res Clin Haematol. 2014; 27: 11–18.
- Mantha S, Tallman MS, Soff GA. What’s new in the pathogenesis of the coagulopathy in acute promyelocytic leukemia?. Curr Opin Hematol. 2016; 23: 121–126.
- Wang P, Zhang Y, Yang H, Hou W, Jin B, et al. Characteristics of fibrinolytic disorders in acutepromyelocytic leukemia. Hematology. 2018; 23: 756–764.
- Kolev K, Longstaff C. Bleeding related to disturbed fibrinolysis. Br J Haematol. 2016; 175: 12–23.
- Kwaan HC, Weiss I, Tallman MS. The Role of Abnormal Hemostasis and Fibrinolysis in Morbidity and Mortality of Acute Promyelocytic Leukemia. Semin Thromb Hemost. 2019; 45: 612–621.
- Avvisati G. Coagulopathy in APL: A step forward?. Blood. 2012; 120: 4–6.
- O’Connell PA, Madureira PA, Berman JN, Liwski RS, Waisman DM. Regulation of S100A10 by the PML-RAR-alphaoncoprotein. Blood. 2011; 117: 4095–4105.
- Liu Y, Wang Z, Jiang M, Dai L, Zhang W, et al. The expression of annexin II and its role in the fibrinolyticactivity in acute promyelocytic leukemia. Leuk Res. 2011; 35: 879–884.
- Wassenaar T, Black J, Kahl B, Schwartz B, Longo W, et al. Acute promyelocytic leukaemia and acquireda-2-plasmin inhibitor deficiency: A retrospective look at the use of epsilon-aminocaproic acid (Amicar) in 30 patients. Hematol Oncol. 2008; 26: 241–246.
- Naymagon L, Mascarenhas J. Hemorrhage in acute promyelocytic leukemia: Can it be predicted and prevented?. Leuk Res. 2020; 94: 106356.
- Rodeghiero F, Avvisati G, Castaman G, Barbui T, Mandelli F. Early deaths and anti-hemorrhagic treatments in acutepromyelocytic leukemia. A GIMEMA retrospective study in 268 consecutive patients. Blood. 1990; 75: 2112–2117.
- Brown JE, Olujohungbe A, Chang J, Ryder WDJ, Morganstern GR, et al. All-Trans Retinoic Acid (Atra) and Tranexamic Acid: A Potentially Fatal Combination in Acute Promyelocytic Leukaemia. Br J Haematol. 2000; 110: 1010–1012.
- Brenner A, Ker K, Shakur-Still H, Roberts I. Tranexamic acid for post-partumhaemorrhage: What, who and when. Best Pr Res Clin Obstet Gynaecol. 2019; 61: 66–74.
- Hibbs SP, Roberts I, Shakur-Still H, Hunt BJ. Post-partum hemorrhage and tranexamic acid: A global issue. Br J Haematol. 2018; 180: 799–807.
- Pacheco L, Hankins GDV, Saad AF, Constantine MM, Chiossi G, et al. Tranexamic acid for the management ofobstetric hemorrhage. Obstet Gynecol. 2017; 130: 765–769.
- Ducloy-Bouthors AS, Duhamel A, Kipnis E, Tournoys A, Prado-Dupont A, et al. Postpartum haemorrhage related early increase in D-dimers is inhibited by tranexamicacid: Haemostasis parameters of a randomized controlled open labelled trial. Br J Anaesth. 2016; 116: 641–648.
- Pabinger I, Fries D, Schöchl H, Streif W, Toller W. Tranexamic acid for treatment and prophylaxis of bleeding and hyperfibrinolysis. Wien Klin Wochenschr. 2017; 129: 303–316.
- WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities inwomen with post-partum haemorrhage (WOMAN): An international, randomised, double-blind, placebo-controlled trial. Lancet. 2017; 389: 2105–2116.
- Ozolina A, Strike E, Jaunalksne I, Krumina A, Bjertnaes LJ, et al. PAI-1 and t-PA/PAI-1 complex potential markers offibrinolytic bleeding after cardiac surgery employing cardiopulmonary bypass. BMC Anesthesiology. 2012; 12: 27.
- Gebhart J, Kepa S, Hofer S, Koder S, Kaider A, et al. Fibrinolysis in patients with a mildto-moderate bleeding tendency of unknown cause. Ann Hematol. 2017; 96: 489–95.