
Special Article: Myelofibrosis
J Blood Disord. 2023; 10(1): 1072.
The Pattern of Thrombosis in Patients with JAK2 V617F Mutation
Riad Akoum, MD*
Medical Oncologist, Lebanese American University Medical Center Rizk Hospital (LAUMCRH), Lebanon
*Corresponding author: Riad Akoum Medical Oncologist, Lebanese American University Medical Center Rizk Hospital (LAUMCRH), Beirut, Lebanon. Email: riad.akoum@laumcrh.com; rakoum@yahoo.com
Received: March 15, 2023 Accepted: April 28, 2023 Published: May 05, 2023
Abstract
Background: JAK2V617F mutation is recurrent in MPN and reported as a marker for occult MPN in patients with splanchnic vein thrombosis.
Objective: To better estimate the pattern of arterial and venous thrombosis in a Lebanese series in order to define the potential risk and to evaluate the current treatment.
Methods: Ninety-five consecutive patients were included and all arterial and venous thrombotic events were documented, altogether with the clinical and demographic data.
Results: Twenty-eight % of patients developed arterial thrombosis and 29% venous thrombosis with PV predilection for DVT and BCS. 27% and 5% of patients had thrombosis at presentation or history of thrombosis. However; subsequent thrombosis may develop in 20% of cases and may be the cause of death in 12.6%.
Two cases with family aggregation were observed.
Conclusion: Close surveillance should be carried out in JAK2V617F-mutated patients with special attention to subsequent thrombosis development. Familial clustering should be looked for.
Abbreviations: SMG: Splenomegaly >5cm from the Costal Margin (SMG in all PMF Patients was >10cm); MDS: Myelodysplastic Syndrome; AML: Acute Myeloid Leukemia; SVT: Splancnic Vein Thrombosis (5 Potal Vein and 1 Budd Chiarri); Mes I: Mesenteric Artery Infarction
Introduction
The Myeloproliferative Neoplasms (MPN), Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) are common hematologic neoplasia, usually affecting individuals over 60 years. The average annual age-adjusted rate is 0.21/100,000 among individuals younger than 40 years and reaches 12.19/100,000 among those older than 80 years [1]. However, due to the relatively smooth clinical course, many cases of PV and ET remain undiagnosed.
Thrombosis, hemorrhage, evolution to more pronounced myelofibrosis and transformation to acute myeloid leukemia are the main complications in the clinical course of ET and PV.
The cumulative risk of thrombosis in PV ranges from 2.5 to 5% per patient-year and from 1.9 to 3% per patient-year in ET [2], the recurrence rate is 5.6% per patient-year in both disorders and may reach 50% at 10 years [3]. Arterial thromboses represent 60% to 70% of events and include Myocardial Infarction (MI), ischemic stroke and peripheral arterial occlusion. Venous thromboses represent 34% to 39% of events in PV and 10% to 29% in ET and include Deep Vein Thromboses (DVT), Pulmonary Embolism (PE), Cerebral Vein Thromboses (CVT) and Splancnic Vein Thromboses (SVT) [4] The thrombotic events represent the initial presentations in up to 39% of PV and ET patients and remain the main subsequent causes of mortality [3]. The fatal complication rate increases with age in PV while life expectancy is not affected in patients with ET [5].
The overall frequency of thrombosis in PMF is 11.6% [6].
PMF is associated with a worse prognosis and marked reduction in life expectancy as compared to PV and ET.
Established risk factors for thrombosis in MPN are age older than 60 and previous thrombotic event. Controversial risk factors are concomitant hypercoagulable state and conventional cardio-vascular risk factor. Recent studies have implicated white blood cell count [7] and JAK2 V617F status [8,9]. Hematocrit level and platelet count, although controversial, have not been associated with the thrombotic event [10,11].
The risk of developing MPN was found to be increased in first degree relatives of patients with MPN [12].
We reviewed retrospectively all patients with MPN treated in our hospital to describe the pattern of thromboses in the Lebanese population in order to evaluate the prevention measures and to estimate the familial aggregation of this disorder.
Study Population
Ninety five consecutive patients with JAK2V617F mutation were included from 2006 to 2019. Clinical and hematological data obtained from patient records and bone marrow biopsies were reviewed. Hematological diagnoses were reconfirmed according to the 2016 WHO criteria. Clinical, hematological features and bone marrow cellular morphology enabled a clear cut distinction between ET, PV, PMF and associated or secondary MF. Arterial and venous thrombotic events included ischemic stroke, MI, angina pectoris, mesenteric artery ischemia, DVT, CVT, SVT and BCS. Stroke was confirmed by computed tomography or brain magnetic resonance, pulmonary embolism was diagnosed by a positive angio-scan or ventilation-perfusion scan, and DVT and SVT were diagnosed by Doppler ultra-sonography or hemodynamic studies. In each patient the thrombotic event occurring at presentation or after diagnosis were recorded. The time from diagnosis to metachronous thrombosis was measured and the ongoing treatment was registered.
Treatment was heterogeneous and consisted of watch-and-wait policy until disease progression, Aspirin, Aspirin +Hydroxyurea, Thalidomide, Ruxolimib, interferon, supportive therapy with blood transfusion and erythropoietin.
Results
Ninety five patients; 49 women and 56 men were analyzed. Table 1 summarizes the main demographic, clinical and laboratory features of the 95 patients at presentation and the main thrombotic events occurring subsequently. At the time of analysis 34 patients had died at a median age of 72 years, 29 of them had primary or concomitant myelofibrosis with ET and PV. The median follow up time was 5.5 years. The cause of death was reported in 30 patients. It was related to thrombotic event in 40% (6 MI, 4 PE, 2 mesenteric artery ischemia), secondary AML in 13.5% and intercurrent disease in 30% of cases. There were one DLBCL, 2 gastro-intestinal hemorrhages, 2 septic choc from intestinal occlusion and 9 non-related diseases. Patients who had thrombosis at presentation had higher risk of dying from thrombotic event. Survival curves are illustrated in figure 1 according to disease phenotype.
![]()
ET
PV
PMF
ET/MF
PV/MF
MDS/MPN
Total (%)
Total (%)
24(25.2%)
36 (37.8%)
19 (20%)
7 (7.3%)
3 (3.1%)
6 (6.3%)
95
Sex F/M
13/11
14/22
9/10
0/7
1/2
2/4
49/56
Mean age
60 [18-90]
53 [24-84]
69 [57-81]
69 [43-77]
70 [59-79]
71 [59-78]
66 [43-77]
History of thrombosis
1
1
5 (5.2%)
Thrombosis at presentation
3
1
1
4
13
26 (27.3%)
Subsequent first thrombosis
3
8
1
4
2
2
20 (20.1%)
Thrombosis; cause of death
5
1
2
4
12 (12.6%)
Thrombosis recurrence
1
4
1
3
9 (9.5%)
AML transformation
-
1
3
1
-
1
6 (6.3%)
Splenomegaly
11 (55%)
16 (46%)
19 (77%)
6 (85.6%)
3 (100%)
6 (100%)
71 (74.2%)
WBC (G/l)
13.4 [6-30]
20.6 [6-40]
10 [1.2-25]
16.3 [6-23]
9 [5-13]
3.5 [1.1-7]
Platelets (G/l)
950 [500-1800]
512 [80-1036]
151 [20-300]
870 [600-1500]
633 [200-1200]
85 [30-130]
First degree relatives with JAK2V617F
-
1
-
-
-
1
2 (2.1%)
Table 1: Patient characteristics, clinical presentation, history of thrombosis and progression.
![]()
ET
PV
PMF
ET/MF
PV/MF
MDS/MPN
Total (%)
Arterial Thrombosis
CVA
-
5
4
1
1
-
11 (11.5%)
MI
3
4
4
2
1
-
14 (14.7%)
Mesenteric Infarction
1
1
-
-
-
-
2 (2.1%)
Venous Thrombosis
DVT
4
11
1
-
1
-
17 (17.9)
PE
-
1
2
-
-
-
3 (3.1%)
SVT
2
2
1
-
1
-
6 (6.2%)
CVT
-
1
-
-
-
1
2 (2.1%)
Table 2: Arterial and venous thrombosis pattern.
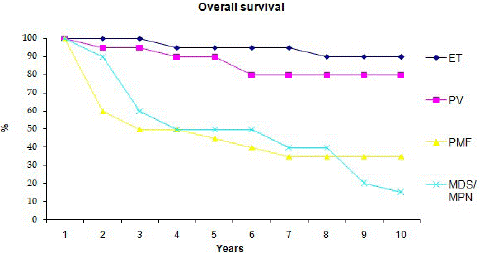
Figure 1: Overall survival of patients with Essential thrombocythemia, Polycythemia Vera, Primary Myelofibrosis and Myeloproliferation +Myelodysplasia.
PMF and myelofibrotic involvement of ET and PV were indistinguishable in phenotype and pattern of molecular abnormalities. Both are characterized by higher rate of Leukemia transformation and shortened OS.
No patient underwent splenectomy or splenic irradiation for symptomatic splenomegaly. No stem cell or bone marrow transplantation was performed.
In 4 patients, the thrombosis occurred within a year period to diagnosis and in 16 other patients within 10 years. Thrombosis was the presenting symptom in 26 patients and occurred subsequently in 18 patients (Table 1).
Non-fatal thrombotic event was present at diagnosis in 21 patients.
The median age of patients who presented with or developed subsequently arterial thrombosis was 65 years where as those who had DVT had a median age of 50 years and those who had PE were elderly patients with a median age of 76 years. There were 6 patients who presented SVT with a median age of 52 years, 3 of them (50%) had previously developed DVT.
There were 16 patients under 40 years of age. All were alive at the time of analysis. Five of them presented with DVT (4 PV and 1 ET). Two of them presented (2PV) with CVA. Two of them developed MI, 3 and 4 years after been diagnosed with ET. A fortuitous hematological testing discovered nine patients.
Investigation of familial clustering in this series has identified a father with PV, completely asymptomatic at 54 years and his 18-year-old daughter with ET who presented with inferior vena cava thrombosis and another 65 years patient with MDS/MPN and his first degree 62 years cousin with PV. No other familial cases of acquired ET and PV were recorded. The presence of thrombophilia conditions was investigated in both patients and turned out to be negative. This investigation included functional protein C, functional antithrombin III, free protein S, activated
PC resistance, lupus anti-coagulant, homocystein, factor V Leiden and factor II mutations.
Leukocytosis at the time of diagnosis defined by a cutoff level of 15 109/l was not correlated with the increased incidence of arterial or venous thrombosis in ET or PMF however it was significantly associated with the occurrence of arterial thrombosis in PV patients (p<0.05) especially MI and CVA.
Discussion
This cohort, hospital-based study was intended to describe the pattern of thrombotic events and the propensity to thrombosis in a Lebanese population with MPN in order to evaluate the prevention measures and to estimate the familial aggregation of this mutation.
JAK2V617F is the most common MPN driver gene mutation and the first molecular diagnostic tool before the emergence of less commonly involved CALR and MPL gene mutations. These mutations activate the physiologic signal-transduction pathways responsible for hematopoiesis.
JAK2V617F acquisition can occur at any age, mainly in women. MPN are uncommon before the age of 50 years but the incidence increases exponentially after the age of 60 years and sometimes associated with PMF and AML and become more common in men [13].
In patients with secondary MF due to PV and ET or PMF, the survival is affected by extra medullary hematopoiesis, marrow failure, and leukemic transformation, targeted therapy with Ruxolitinib, pegylated interferon, bone marrow transplantation or epigenetic-modifying drugs are used. However; in patients with PV and ET the cornerstone of therapy is lowering the cell mass and preventing platelet aggregation.
The morbidity and the mortality of PV and ET are severely affected by arterial thrombosis, venous thrombosis at atypical sites and microcirculatory disturbances.
Although patients with MPN across all age groups are at significantly increased rate of arterial and venous thrombosis compared to matched population control [14], the development of thromboembolic disease is multifactorial. A single patient may have multiple underlying conditions. Venous thrombosis may be potentiated by orthopedic or oncologic surgery, exogenous estrogen and immobilization. Arterial thrombosis may be potentiated by smoking, obesity, hyperlipidemia, diabetes and hereditary predisposition to hypercoagulability.
The cumulative rate of thrombosis was 55.8%. Fifty two% were arterial, 37% were venous and 11% involved the splanchnic veins. Thrombosis was the presenting symptom in 28% of cases. Fatal thrombotic events represented 22% of all thromboses, mainly due to MI, PE and CVA.
Age and history of thrombosis are the main risk factors for thrombosis in PV and ET [2,4]. Leukocytosis [15] and JAK2 mutation [16], which has also been reported as risk factors are potentially modifiable with treatment. Cellular and molecular analysis have attributed the hypercoagulable state generated by ET and PV to the activated neutrophils and platelets that contribute to the acquired activated protein C resistance associated with low free protein S levels [17].
There is a state of overlapping between the hematological manifestations of PV, ET and PMF which may represents diagnostic challenges. JAK2 V617F-positive cases share many clinical and laboratory features. Therefore, these disorders may be considered as parts of a phenotypic continuum. [18,19]. Only erythrocytosis distinguishes PV from ET and PMF, ET does not have a unique clinical phenotype because isolated thrombocytosis can also be the presenting feature of PV and PMF. PMF with excess in platelet count called false ET is distinguishable from ET by bone marrow histopathologic and morphologic studies. PMF and fibrotic transformation of ET and PV are indistinguishable in terms of phenotype and pattern of molecular abnormalities and both are characterized by higher rate of leukemic transformation and shortened overall survival [5]. In most studies, PMF has been shown to have a comparable thrombotic profile with ET but much lower than PV.
Splanchnic Vein Thrombosis (SVT) includes Portal Vein Thrombosios (PVT), Mesenteric Vein Thrombosis (MVT) and Budd Chiari Syndrome (BCS). Hormonal treatment is a potent associated risk factor to PV in BCS and liver cirrhosis, Inflammatory Bowel Diseases (IBD), abdominal cancer and surgery are the most common risk factors in PVT and MVT [20].
Six patients in our series had SVT. The presence of JAK2V617F mutation has been reported in patients with SVT even in the absence of MPN at diagnosis [21]. Its presence is highly associated with SVT. Screening for the mutation in this condition is recommended. However, routine testing for is not recommended for patients with first episode venous thrombosis at a usual site or solitary arterial thrombosis [22].
BCS and/or PVT are frequent presenting features of MPN, particularly in young patients. The classical features of MPN could be masked in PVT and BCS by consequences of portal hypertension; splenomegaly, hemodilution and variceal bleeding. Recent meta-analysis has estimated the prevalence of JAK2 mutation in BCS and PVT at 32.7% and less than 2.57% in venous thrombosis [23]. Our series contains 6 PVT, 2 of them caused by ET and 4 of them by PV, 2 males and 4 females with a mean age of 57 years and only 2 young female patients with PV causing BCS.
Only 2 first-degree relatives were identified in our series (2.1%). A father with PV and his daughter with ET and another patient with MDS/MPN and his first degree cousin with PV. Cases with family history of thrombocythemia were detected at a young age and have been associated with somatic JAK2 V617F mutation [24] and germline JAK2 V617I alteration [25].
In familial MPN, the JAK2 mutation is acquired and occurs as secondary genetic event [26]. The presence of two patients in a same pedigree with mutations of the JAK2 gene, suggests that a genetic predisposition to the acquisition of the JAK2 mutations is supposed to be inherited. The prevalence of familial cases within MPN was estimated at 7.6% and the inheritance pattern was consistent with an autosomal dominant trait with decreased penetrance. The clinical presentation at diagnosis is similar in familial and sporadic cases [26]. In a population based study by Nielson et al [27] 49.488 individuals from Copenhagen had been tested for JAK2V617F, 63 (0.1%) were positive, among them 48 developed eventually MPN.
Controversial reports had implicated the leukocytosis as an adverse prognostic factor in terms of thrombosis [28,29]. Our series comprised low and high-risk patients.
The SVT, Portal Vein Thrombosis (PVT), mesenteric thrombosis, hepatic vein thrombosis or Budd Chiari syndrome (BCS) have higher prevalence among patients with MPN than patients with other primary hypercoagulable state [29]. The JAK2 V617F mutation has been found in 45% and 34% of blood samples from patients with BCS and PVT respectively indicating a strong association between MPN and SVT [23].
PMF is a heterogeneous disease in terms of presentation and evolution characterized by constitutive association of reticulin or collagen in bone marrow with megakaryocytic hyperplasia or dysplasia and mobilization of hematopoietic progenitor cells with extramedullary hematopoiesis. Patients may present with high, normal or low platelet and white blood cell counts. They may remain asymptomatic for long periods of time or have symptoms related to anemia and splenomegaly. Although a significant increase in venous thrombosis was noted in PMF as compared with the general population, thrombotic events occur less commonly and hemorrhage occurs more commonly in PMF than PV and ET. Factors associated with thrombosis in PMF are thrombocytosis, cellular phase which is the myeloproliferative form of the disease and cardiovascular risk factors. In addition, advanced age, anemia, RBC transfusion dependency, leucopenia, leukocytosis, thrombocytopenia, peripheral blast count, systemic symptoms, degree of micro vessel density and cytogenetic abnormalities were shown to be associated with poor outcome in patients with PMF. The presence of JAK2V617F mutation per se does not seem to imply worse survival. [5].
The true incidence of MPN underlying arterial and/or venous thrombosis may be underestimated because of the great number of patients who have concomitant cardiovascular risk factors, secondary polycythemia related to tobacco use or blood cytopenia related to age and because of the occult forms that may pass unnoticed.
It has been suggested that patients with MPN are prone to the development of second cancer, especially those experiencing arterial events after MPN diagnosis [31]. Our series highlight the development of a diffuse large B-cell lymphoma in a patient with PMF and the emergence of a chronic lymphocytic leukemia in a patient with PV after 10 years of evolution.
Concomitant emergence of bcr-abl CML in the setting of JAK2V617F has also been described [32]. One elderly patient in our cohort has concomitantly CML
MDS/MPN overlap syndromes with JAK2V617F were seen in 6 of our patients, 4 MDS/MPN with ring sideroblasts, one unclassified MDS/MPN and one CMML.
References
- Rollison DE, Howlader N, Smith MT. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood. 2008; 112: 45-52.
- Marchioli R, Finazzi G, Landolfi R. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005; 23: 2224-32.
- De Stefano V, Za T, Rossi E. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. 2008; 93: 372-80.
- Carobbio A, Thiele J, Passamonti F. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011; 117: 5857-9.
- Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia. 2008; 22: 905-14.
- Cervantes F, Alvarez-Larrán A, Arellano-Rodrigo E. Frequency and risk factors for thrombosis in idiopathic myelofibrosis: analysis in a series of 155 patients from a single institution. Leukemia. 2006; 20: 55-60.
- Barbui T1, Carobbio A, Rambaldi A. Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: is leukocytosis a causative factor? Blood. 2009; 114: 759-63.
- Dahabreh IJ, Zoi K, Giannouli S. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res. 2009; 33: 67-73.
- Vannucchi AM, Antonioli E, Guglielmelli P. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. 2008; 22: 1299-307.
- Di Nisio M, Barbui T, Di Gennaro L, Borrelli G, Finazzi G, et al. The haematocrit and platelet target in polycythemia vera. Br J Haematol. 2007; 136: 249-59.
- Tefferi A, Gangat N, Wolanskyj AP. Management of extreme thrombocytosis in otherwise low-risk essential thrombocythemia; does number matter? Blood. 2006; 108: 2493-4.
- Landgren O, Goldin LR, Kristinsson SY. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood. 2008; 112: 2199-204.
- Spivak JL. Myeloproliferative Neoplasms. N Engl J Med. 2017; 377: 895-6.
- Hultcrantz M, Björkholm M, Dickman PW. Risk for Arterial and Venous Thrombosis in Patients With Myeloproliferative Neoplasms: A Population-Based Cohort Study. Ann Intern Med. 2018; 168: 317-325.
- Barbui T1, Carobbio A, Cervantes F. Thrombosis in primary myelofibrosis: incidence and risk factors. Blood. 2010; 115: 778-82.
- Lussana F, Caberlon S, Pagani C. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009; 124: 409-17.
- Marchetti M, Castoldi E, Spronk HM. Thrombin generation and activated protein C resistance in patients with essential thrombocythemia and polycythemia vera. Blood. 2008; 112: 4061-8.
- Campbell PJ1, Scott LM, Buck G. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005; 366: 1945-53.
- Spivak JL. Narrative review: Thrombocytosis, polycythemia vera, and JAK2 mutations: The phenotypic mimicry of chronic myeloproliferation. Ann Intern Med. 2010; 152: 300-6.
- Finazzi G, De Stefano V, Barbui T. Splanchnic vein thrombosis in myeloproliferative neoplasms: treatment algorithm 2018. Blood Cancer J. 2018; 8: 64.
- De Stefano V, Fiorini A, Rossi E. High prevalence of the JAK2 V617F mutation in patients with extrahepatic portal vein thrombosis. Hepatology. 2007; 45: 831-2.
- Xavier SG, Gadelha T, Rezende SM. JAK2V617F mutation in patients with thrombosis: to screen or not to screen? Int J Lab Hematol. 2011; 33: 117-24.
- Dentali F, Squizzato A, Brivio L. JAK2V617F mutation for the early diagnosis of Ph- myeloproliferative neoplasms in patients with venous thromboembolism: a meta-analysis. Blood. 2009; 113: 5617-23.
- Bellanné-Chantelot C, Rabadan Moraes G, Schmaltz-Panneau B. Germline genetic factors in the pathogenesis of myeloproliferative neoplasms. Blood Rev. 2020; 42: 100710.
- Mead AJ, Rugless MJ, Jacobsen SE. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med. 2012; 366: 967-9.
- Rumi E. Familial chronic myeloproliferative disorders: the state of the art. Hematol Oncol. 2008; 26: 131-8.
- Nielsen C, Bojesen SE, Nordestgaard BG. JAK2V617F somatic mutation in the general population: myeloproliferative neoplasm development and progression rate. Haematologica. 2014; 99: 1448-55.
- Gangat N, Strand J, Li CY. Leucocytosis in polycythaemia vera predicts both inferior survival and leukaemic transformation. Br J Haematol. 2007; 138: 354-8.
- Gangat N, Wolanskyj AP, Schwager SM. Leukocytosis at diagnosis and the risk of subsequent thrombosis in patients with low-risk essential thrombocythemia and polycythemia vera. Cancer. 2009; 115: 5740-5.
- Kiladjian JJ, Cervantes F, Leebeek FW, Marzac C, Cassinat B, et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008; 111: 4922-9.
- De Stefano V, Ghirardi A, Masciulli A. Arterial thrombosis in Philadelphia-negative myeloproliferative neoplasms predicts second cancer: a case-control study. Blood. 2020; 135: 381-386.
- Lorenzo M, Grille S, Stevenazzi M. Emergence of BCR-ABL1 Chronic Myeloid Leukemia in a JAK2-V617F Polycythemia Vera. J Hematol. 2020; 9: 23-29.