
Research Article
J Blood Disord. 2024; 11(2): 1088.
Complications of Hospitalized Children with Sickle Cell Anemia from A Referral Center in Southern Nigeria
Bassey EU¹*; Akpan IS²; Iyanam VE³; Nnoli C¹
¹Department of Pediatrics, Faculty of Clinical Sciences, University of Uyo, Nigeria
²Department of Hematology, Faculty of Clinical Sciences, University of Uyo, Nigeria
³Department of Family Medicine, Faculty of Clinical Sciences, University of Uyo, Nigeria
*Corresponding author: Eno-Obong U Bassey Department of Paediatrics, Faculty of Clinical Sciences, University of Uyo, Uyo. Nigeria. Tel: +234 806 408 7511 Email: utukenoobong@yahoo.com
Received: August 28, 2024 Accepted: September 19, 2024 Published: September 26, 2024
Abstract
Introduction: Sickle cell anaemia is an inherited blood disorder widely prevalent in the west African sub-region. Children born with this disorder experience frequent episodes of pain and other morbidities requiring hospitalization. Knowledge of the medical complications which commonly present in various regions and centers is important, as it can help clinicians with prompt identification of symptoms, and institution of relevant medical care and treatment. This would greatly benefit outcome.
Methods: A retrospective study, done over a thirty-month period, from January 2021 to June 2023. Information was retrieved from the case folders of hospitalized children with sickle cell anaemia in the Paediatric Medical ward. These included the biodata, medical history, physical examination and results of relevant haematological, radiological or other investigations. Retrieved data was entered into an excel spreadsheet. Analysis was done using the Microsoft excel package and variables were expressed in percentages, tables and figures.
Results: Total ward admissions during the study period were 1282. Of these, 131 were children admitted for various complications of sickle cell anaemia. These constituted 10.2% of the total admissions, with a male/ female ratio of 1:1. The causes of hospital admissions in decreasing frequency were vaso-occlusive crises (48.1%), infection (7.6%) and anaemia (5.3%) respectively. Children less than ten years of age had a greater percentage of hospital admissions. No child died within this study period from complications of sickle cell anaemia.
Conclusion: Vaso-occlusive crises, infection and anaemia were the commonest causes of hospital admission among children with sickle cell anaemia in this center. The outcome of hospital admissions in these children were good, with most being discharged in satisfactory condition. Comprehensive care for these children is essential for good outcome.
Keywords: Children; Complications; Sickle cell anaemia
Introduction
Sickle cell anaemia is an inherited genetic disorder prevalent in the sub-Saharan African region. It represents a major public health problem and a burden to affected children, families, healthcare system and the society at large. It is a disease fraught with repeated episodes of acute illnesses and frequent hospitalizations since it causes a wide range of multisystemic complications and progressive organ damage [1,2]. It is important for clinicians practicing in countries where sickle cell anaemia is prevalent to be aware of common presenting complications of this illness in their locality. This would heighten their index of suspicion in diagnosis and treatment. This review is aimed at highlighting such complications. It is also needful to generate more data in different localities about the incidence and prevalence of various sickle cell complications observed in the childhood population. Health-care policy makers would then be alerted on the need for more focused and targeted interventions in the comprehensive care of these children. These would improve the long-term survival of the child, and enhance their quality of life.
Methods
A retrospective study, done over a thirty-month period, from January 2021 to June 2023. Information retrieved from the case folders of hospitalized sickle cell anaemic children in the Paediatric Medical ward, included the biodata, history, physical examination and results of relevant haematological, radiological or other investigations done. Retrieved data was entered into an excel sheet. Analysis was done using the Microsoft excel package and variables were expressed in percentages, tables and figures.
Results
Total ward admissions during the study period was 1282. Of these, 131 were children admitted for various complications of sickle cell anaemia. These constituted 10.2% of the total admissions, with a male/female ratio of 1:1 The age range of children was between 8 months to 17 years, with a median age of 8 years. Children ten years of age and below, were the most hospitalized group - 63% (Table 1).
![]()
Age (years)
N� (%)
<5
43 (33)
6�10
40 (30)
11-15
38 (29)
>15
10 (8)
TOTAL
131 (100.0)
Table 1: Age range of hospitalized children.
![]()
Variable
Number (n)
Vaso-Occlusive Crisis (Bone-pain)
63
Sepsis (Infection)
10
Dactylitis
7
Severe anaemia
7
Acute chest syndrome
5
Bronchopneumonia
5
Lobar pneumonia
5
Anaemic heart failure
4
Stroke
4
Osteomyelitis
4
Severe Malaria
3
Urinary tract infection
2
Chronic leg ulcer
2
Septic arthritis
2
Retinopathy
2
Nephropathy
2
Meningitis
1
Carvenous sinus thrombosis
1
Avascular necrosis of the femoral head (ANFH)
1
TOTAL
131
Table 2: Spectrum of complications in hospitalized sickle cell anaemic children.
The three leading causes of hospitalization were vaso-occlusive Crisis (48.1%), infection (7.6%) and hyper-haemolytic anaemia (5.6%). Respiratory complications also played a major role in hospitalization of children. Central nervous system complications like meningitis and carvenous sinus thrombosis were the least frequent causes of admissions. No patient died from complications of sickle cell anaemia within the study period.
The age group with the highest frequency of vaso-occlusive (bone-pain) crisis were the older children in the 11 to 15-year group, followed by children less than five years old (Figure 1).
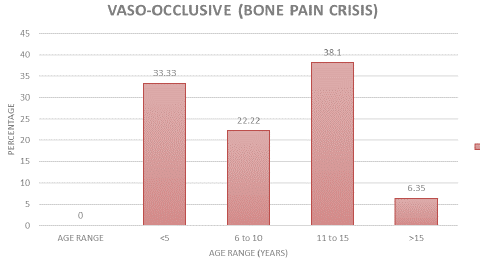
Figure 1: Age grade of children with Vaso-Occlusive cris.
The systemic profile of various complications seen (Figure 2) illustrated the following in decreasing frequency: Musculo-skeletal complications (n =16) which included dactylitis, chronic leg ulcers, osteomyelitis, septic arthritis, avascular necrosis of femoral head. Respiratory complications (n =15); acute chest syndrome, bronchopneumonia, lobar pneumonia. Cardiovascular complications (n=11); anaemic heart failure and severe anaemia. Central nervous system complications (n =10): These included stroke, carvenous sinus thrombosis, meningitis, sickle-cell retinopathy. Renal complications (n=4): urinary tract infection and sickle cell nephropathy respectively.
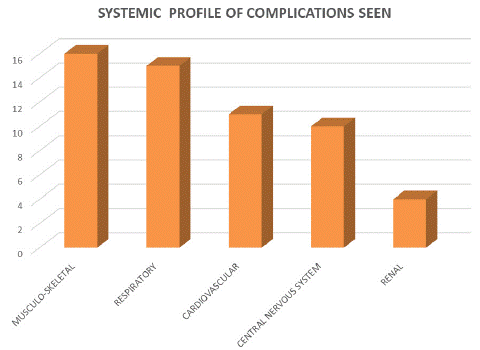
Figure 2: Systemic profile of complications seen.
Figure 3 illustrates the outcome of the children hospitalized. A greater percentage of the admitted children (94.7%) were discharged in satisfactory condition while a few (5.3%) Left Against Medical Advice (LAMA). No child with sickle cell anaemia absconded and none died during the period of study.
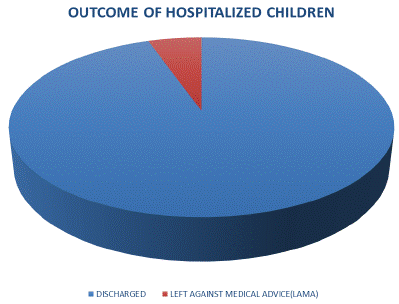
Figure 3: Outcome of hospitalized sickle cell children.
Discussion
Sickle Cell Anaemia (SCA) is a chronic haemolytic blood disorder, which is inherited in an autosomal recessive manner. SCA is a recognized global public health concern due to the significant social and economic impact that results from its attendant morbidity [1,2]. Studies from other African countries have similar observations to present report, that common morbidities associated with sickle cell anaemia in children include intermittent painful crisis, musculoskeletal problems, stroke, pulmonary hypertension and septicaemia [1-4]. These often co-exist, and affect child’s quality of life.
The systemic and life-threatening complications that occur in children with sickle-cell anaemia basically results from acute sickling of red blood cells and sequestration in various organ/systems culminating in the various presentations. If left untreated, could lead to death. It is therefore important for clinicians to recognize these morbidities early and institute appropriate interventions for long-term survival.
The clinical manifestations of sickle cell anaemia for affected children are variable. The bases for this are poorly understood, but it is generally accepted that an interplay of both genetic and environmental factors contribute to the varying clinical course of the disease. The environmental and economic factors differ in different regions of the world where these children live, and inevitably affects their disease course and presentations [1-4].
Within the study period, children admitted with various complications of sickle cell anaemia constituted 10.2% of the total paediatric medical admissions. This certainly represents a great burden from a non-communicable disease. This is comparable to the 10.0% documented in a recent report by Ibraheem et al [5] from ten tertiary hospitals across five geo-political zones in Nigeria, but differs from the lower prevalences of paediatric sickle cell admissions documented from Enugu [6], south-east Nigeria and Port-Harcourt [7], south-south Nigeria, recording 3.9% and 3.7% respectively of total paediatric medical admissions. The erstwhile study in Enugu was done some years ago, of which presently there is more awareness on sickle cell disease among communities and families in Nigeria. Nevertheless sickle cell anaemia constitutes a huge burden to childhood paediatric hospitalizations in sub-Saharan Africa, and comprehensive care with interventions must necessarily be considered in health care policy decisions of several African countries, including Nigeria. Children less than ten years of age were the most hospitalized group comparable to other reports from Nigeria and other countries [7-15].
Vaso-occlusive (bone-pain) crisis, infections, anaemia and pneumonias were seen as the most frequent causes of hospital admissions among the children with sickle cell anaemia in this study. Similar trend of complications has been documented by various authors within and outside Nigeria in hospitalized sickle cell children [11-16]. The disease process in sickle cell anaemia causes complications in multiple organ/systems of these children. These often co-exist affecting quality of life, and without prompt intervention could progress to death.
Acute bone-pain crisis (vaso-occlusive) was the most common cause of hospital admission in this study. This observation is similar to reports from Nigeria and other countries [9,13-16]. In contrast some other reports from Southern Nigeria documented infection a leading cause of hospital admissions [6,8]. Pain associated with vaso-occlusive crises is due to occlusions of microvascular vessels, which trigger the activation of nociceptive afferent nerve fibers [17,18]. Children younger than 3 years are prone to painful swelling of the hands, known as hand-foot syndrome or dactylitis. Older children and young adolescents had the greatest frequency of vaso-occlusive crises in present study. A similar observation to a report from Port-harcourt, Nigeria where a higher frequency of children greater than 12 years of age were the most affected [8]. Long bones and joints are often the affected areas of necrosis leading to pain [17,18].
Infections were the second-leading cause of hospitalization among children and adolescents in this study. Other studies had also noticed similar trend, with infection being a second leading cause of hospital admissions [8,15,17,19]. A major cause of mortality in childhood sickle cell anaemia, is overwhelming bacterial infections especially due to encapsulated organisms, principally pneumococcus [17,18]. This is as a result of a variety of immune defects in the disease most importantly, splenic dysfunction. Most of these children become functionally asplenic early in life [17,18]. The risk of bacterial infection is dramatically increased in sickle cell anaemia and especially so, for encapsulated organisms and children less than five years. This is due to repeated episodes of sickling-induced splenic infarction. Additional contributing factors include abnormalities of opsonization, antibody production, the alternate complement pathway, leucocyte function defects, and cell-mediated immunity [17,18].
Anaemia is a major cause of morbidity and mortality in SCD, and many patients die in hospital emergency rooms and wards before blood transfusions can be initiated. Authors within and outside Nigeria have noted similar picture. Most cases were related to aplastic, acute sequestration, hyper-hemolytic and vasculo-occlusive crises [6,11]. The authors suggested, however, that it was important to assess for other causes of anaemia in SCA patients including malaria parasitaemia which is also known to be an incriminating factor for anaemia in these children [6,11,19].
The systemic profile of complications in present study revealed a high frequency of children presenting with musculo-skeletal complications. Some other authors have noted similarly, that many children with sickle cell anaemia experience musculoskeletal complications due to avascular necrosis, osteomyelitis, and septic arthritis [20-22]. The musculoskeletal complications of SCA result from vessel occlusion, tissue ischemia, infarction and progressive end organ damage. These authors noted that children below age 10 years were more likely to have multiple musculoskeletal problems [20-22].
Respiratory complications such as pneumonias and acute chest syndrome, also presented a great number of total admissions. A multi-center study across several regions in Nigeria by Ibraheem et al recorded respiratory morbidities in hospitalized sickle cell children to include pneumonia (40.1%) and acute chest syndrome (26.7%) respectively [5]. Central nervous system complications like stroke and multiple organ failure are other common causes of morbidity and death [5].
In older patients, chronic organ failure, especially renal failure, becomes quite important. Some other distressing, but not necessarily fatal complications include chronic leg ulcers and avascular bone necrosis typically affecting the femoral heads also occurring especially in older children and adolescents. All these were documented with differing frequencies in children seen over the study period.
Most of the children were discharged in better condition, and no deaths were recorded during this study period. There has been widespread improvement in the management of children born with sickle cell anaemia over the recent years. This has led to the prolonged lifespan of affected children to late adulthood and beyond. The decrease in mortality among SCA children even in low-resource countries is partly attributed to the implementation of comprehensive care programs in many centers. These comprehensive practices encompass, health education in communities, and the general public, childhood immunizations and vaccinations, anti-malarial prophylaxis therapy, vitamin supplements and patient/caregiver empowerment through education [23-25]. The health education addresses helpful practices such as the importance of hydration, adequate nutrition, and other strategies that help prevent the onset of acute SCA complications. These practices have been adopted in this study center and may be greatly contributory for the favourable outcome observed in the study period. Although somewhat encouraging, the relatively low in-hospital mortality among SCA children may also be under-reporting the true mortality from the disease considering community deaths and deaths that could occur before SCA diagnosis is made [18,23-25].
Conclusion
Vaso-occlusive crisis, infections and severe anaemia were the most frequent causes of hospital admissions among the children with sickle cell anaemia. There was a good outcome for discharge among most of the patients with no death recorded. The introduction and use of early (neonatal) screening methods for this disorder, and the sustained use of comprehensive interventions directed at prevention of SCA complications already incorporated in the country, such as antimalarial prophylaxis and immunizations, will help reduce significantly the frequency of hospitalizations and subsequently, the economic burden of the disease. Sustained health education to families, especially those with an affected member would improve health-care seeking behaviours. Government health policies for comprehensive care would be highly beneficial, including increased funding for sickle cell anaemia programs
References
- Mulumba L, Wilson L. Sickle cell disease among children in Africa: An integrative literature review and global recommendations. Int J Nursing Sci. 2015; 3: 56-64.
- Serjeant GR. The clinical features of sickle cell disease. In: Higgs D, Weatherall DJ (Eds). The Haemoglobinopathies: Bailliere’s Tindall, London, United Kingdom. 1993; 93-115.
- Embury SH, Steinberg MH. Clinical Considerations. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH (Eds). In: Sickle Cell Disease: Basic Principles and Clinical Practice, Raven Press, N.Y. 1994: 505-734.
- Abd El-Ghany SM, Tabbakh AT, Nur KI, Abdelrahman RY, Etarji SM, Almuzaini BY. Analysis of causes of hospitalization among children with sickle cell disease in a group of private hospitals in Jeddah, Saudi Arabia; J Blood Med. 2021; 12: 733-740.
- Ibraheem RB, Abdulkadir MB, Aliu R, Issa A, Ibrahim OR, Bello AO et al. Burden and outcome of respiratory morbidities among children and adolescents with sickle cell disease – A retrospective review of emergency presentations in some Nigerian institutions. Plos ONE. 2024; 19: e303323.
- Ikefuna AN, Emodi IJ. Hospital admission of patients with sickle cell anaemia pattern and outcome in Enugu area of Nigeria. Niger J Clin Pract. 2007; 1: 24–29.
- George IO, Opara P. Sickle Cell Anaemia: A survey of associated morbidity in Nigerian Children. Medicine 2011; 7.
- West BA, Aitafo JE. Prevalence, Pattern of disease and outcome of children with sickle cell disease admitted in a private health facility in southern Nigeria. Asian J Pediatr Res. 2023; 12: 17-27.
- Akar NA, Adekile A. Ten-year review of hospital admissions among children with sickle cell disease in Kuwait; Medical Principles and Practice. 2008; 17: 404–408.
- Hagins H, Aboagye RG, Aboye MB, Abu-Gharbieh E, Abu-Zaid A, Addo IY et al. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023; 10: e585–99.
- Chimbatata CS, Chisale MR, Kayira AB, Sinyiza FW, Mbakaya BC, Kaseka PU, et al. Paediatric sickle cell disease at a tertiary hospital in Malawi: a retrospective cross-sectional study. BMJ Paediatr Open. 2021; 5: e1097.
- Oppong-Mensah YG, Odoom SF, Nyanor I, Paintsil V, Amusu EX, Yawnumar SA, et al. Hospitalizations among children with sickle cell disease enrolled in the Kumasi Cell Pan African Consortium (SPARCo) database: A cross sectional study. Health Sci Rep. 2023; 6: e1534.
- Salman ZA, Hassan MK. Hospitalization Events among Children and Adolescents with Sickle Cell Disease in Basra, Iraq. Anemia. 2015; 2015: 195469.
- Jaiyesimi F, Pandey R, Bux D, Sreekrishna Y, Zaki F, Krishnamoorthy N. Sickle cell morbidity profile in Omani children. Annals Trop Paediatr. 2002; 22: 45–52.
- Brown BJ, Jacob NE, Lagunju IA, Jarrett OO. Morbidity and mortality pattern in hospitalized children with sickle cell disorders at the University College Hospital, Ibadan, Nigeria. Niger J Paed. 2013; 40: 34–39.
- Sendy JS, Alsadun MS, Alamer SS, Alazzam SM, Alqurashi MM, et al. Frequency of Painful Crisis and Other Associated Complications of Sickle Cell Anemia Among Children. Cureus. 2023;15: e48115.
- Serjeant GR. Mortality from sickle cell disease in Africa. BMJ. 2005; 330: 432-3.
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004; 364: 1343–1360.
- Utuk EE, Akpan MU. The pattern of morbidity in children with sickle cell anaemia at the University of Uyo Teaching Hospital, Uyo. Akwa Ibom State, Nigeria. Int J Health Sci Res. 2015; 5: 91-97.
- Balogun RA, Obalum DC, Giwa SO, Adekoya-Cole TO, Ogo CN, Enweluzo GO. Spectrum of musculo-skeletal disorders in sickle cell disease in Lagos, Niger J Orthop Surg Res. 2010; 5: 1749-799.
- Chinawa JM, Chukwu BF, Ikefuna AN, Emodi IJ. Musculoskeletal Complications Among Children with Sickle Cell Admitted in University of Nigeria Teaching Hospital Ituku - Ozalla Enugu: A 58 Month Review. Ann Med Health Sci Res. 2013; 3: 564–567.
- Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol. 2005; 129: 482–90.
- Galadanci N, Wudil BJ, Balogun TM, Ogunrinde GO, Akinsulie A, Hasan-Hanga F, et al. Current sickle cell disease management practices in Nigeria. Int Health. 2014; 6: 23–28.
- Nnodu OE, Oron AP, Sopekan A, Akaba GO, Piel FB, Chao DL. Child mortality from sickle cell disease in Nigeria: a model estimated, population-level analysis of data from the 2018 Demographic and Health Survey. The Lancet. com/haematology. 2021; 8: e723-e731.
- Rahimy MC, Gangbo A, Ahouignan G, Adjou R, Deguenon C, Goussano S, et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anaemia in a sub-Saharan African setting. 2003; 102: 834-83.