
Review Article
J Blood Disord. 2015;2(1): 1023.
Inflammation and Coagulation: A “Continuum” Between Coagulation Activation and Prothrombotic State
Guido D’Angelo*
Laboratorio di Chimica-Clinica, Ematologia e Microbiologia Azienda Ospedaliera “S. Antonio Abate” Gallarate Varese, Italy
*Corresponding author: Guido D’Angelo,(Ematologia/Coagulazione) Laboratorio di Chimica-Clinica, Ematologia e Microbiologia, Azienda Ospedaliera“S. Antonio Abate” – Gallarate Via Pastori 4, Varese,Italy.
Received: February 20, 2015; Accepted: March 11, 2015; Published: March 19, 2015
Abstract
The close relationship between inflammation and coagulation is expressed mainly in three stages: blood coagulation activation, a decrease in natural anticoagulants activity and fibrinolytic process decrease. The connection between inflammation and coagulation is now well established. The inflammation also acts on the antithrombotic endothelial normal functions. The loss of homeostasis in normal coagulation processes, promotes not only a stressed thrombotic condition, but it leaves the inflammatory process constantly turned on.
The inflammation/coagulation interaction could be represented like “a dog that bites its tail”.
The review also considers the main coagulation testing and some therapeutic indications. The latter, although experimental and more expensive, in severe sepsis can replace the deficiency of natural anticoagulants, as well as reduce the inflammatory state.
Keywords: Inflammation; Coagulation; Prothrombotic state; Endogen anticoagulant; Fibrinolysis; Coagulation tests
Introduction
The severe and systemic inflammatory response may cause significant pathological damage. There is a thin line between inflammation and coagulation system, and often coagulation disorders are associated with inflammatory diseases.
In severe inflammation, the onset of coagulation disorder is due to the imbalance between coagulation and fibrinolysis activity, with a consequent hypercoagulable state. The outcome is a thrombotic condition with microvascular failure and the development of Multiple Organ Dysfunction Syndrome (MODS) [1-3].
Inflammation and coagulation
The activated coagulation, the endogenous anticoagulants down-regulation and the fibrinolysis inhibition, are the three main mechanisms by which inflammation occurs in the clotting process. During inflammation, the cytokines, which are immune system mediators, are released. The release of cytokine Interleukin-6 (IL- 6) stimulates the production of platelets [4]. In addition, several inflammatory mediators, such as bacterial endotoxins, thromboxane A2, Platelet Activating Factor (PAF) and the neutrophil cathepsin G enzyme, activate the platelets [5]. The production and activation of processes mediated by inflammation give thrombogenic characteristics to the platelets, resulting in increased sensitivity to platelet agonists. Platelet activation induced by inflammation can trigger a loop that perpetuates the inflammatory state through two conditions. In the first case, the activated platelet aggregate make the phospholipid surfaces able to trigger the secondary hemostasis by acquiring negative charge (Figure 1). The resulting production of Thrombin binds to specific cell surface receptors known as “Protease- Activated Receptor” (PAR) and is itself a strong inflammation mediator [6]. In the second case, the interaction between activated platelets and sub endothelial cells stimulates the adhesion and the recruitment of inflammatory leukocytes that release the cytokine Interleukin-1β (IL-1β), which is able to enhance the adhesion properties of endothelial cells [7,8] (Figure 1).
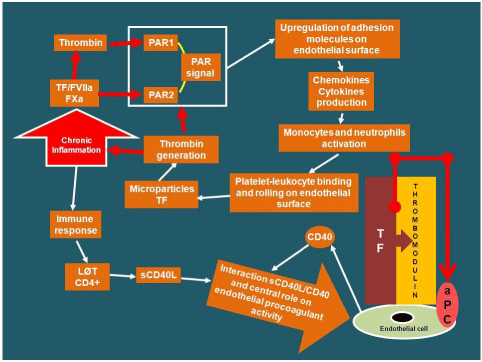
Figure 1: Relationship between inflammation and coagulation. Role of
immune response induced by inflammation on endothelium procoagulant
activity. (TF: Tissue Factor; PAR: Protease-Activated Receptor; LØT:
Lymphocytes T; sCD40L: soluble CD40 Ligand; aPC: activated Protein C).
Other inflammatory mediators, such as Tissue Factor Necrosis-a (TNF-a) and others inflammatory cytokines, or lipoproteins, C-reactive protein and bacterial endotoxins, involving the Tissue Factor (TF), have a procoagulant effect until the formation of thrombin [9-12].
Independently from TF involvement, the monocytes, even by only inflammatory activation, may trigger the coagulation process; they directly activate FX with thrombin generation [13].
There is a close correlation between inflammatory mediator’s release, as well as immune system, and endothelial surface molecules modulation. This results in abnormal endothelial prothrombotic activity, endothelial fibrin deposition, and Thrombomodulin downregulation [14]. Normally, Thrombomodulin-Thrombin link ensure a continuous antithrombotic endothelium function activating Protein C (PC) [15]. In inflammatory stimulus, the activated endothelial cells express transiently Tissue Factor (TF) that, linked to FVIIa, activates both extrinsic and intrinsic coagulation pathways [16] (Figure 2). The mediation between endothelial Thrombomodulin and TF can occur through T-cells CD4+ activation that, stimulated by inflammatory immune-mediated response, produce soluble CD40 ligand (sCD40L). The interaction between sCD40L and CD40 (the latter produced by endothelial cells) plays a central role on endothelium procoagulant activity. In fact, the sCD40L and CD40 interaction down-modulates the Thrombomodulin expression on endothelial cells inducing TF production. The TF-FVIIa link continuous the coagulative activity and consequently, on endothelial surface the fibrin deposition [17,18] (Figure 1). Patients with unstable angina show increased blood coagulation pathway activation and sCD40L up-regulation [19]; consequently, in case of plaque rupture coronary thrombosis may occur [20,21].
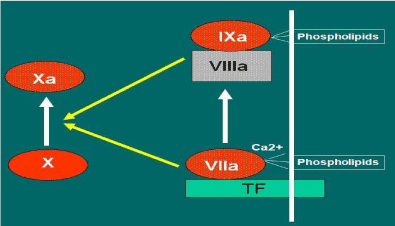
Figure 2: Activation of the coagulation process by the central role of the link
“Tissue Factor (TF) -FVIIa”.
Endogen anticoagulants down-regulation
The expression and function of Antithrombin (AT) and activated Protein C (aPC) decrease with inflammatory process, while the Tissue Factor Pathway Inhibitor (TFPI) concentration may not be abnormal. In inflammation, as the aminoglycanheaparin-like on the endothelial surface are decreased, the AT functional activity may be < 50% [22].
The inhibition of both Thrombomodulin and Endothelial Cell Protein C Receptor (EPCR) transcription determines the aPC decrease (Figure 3). Not only the hepatic deficit synthesis, but also the increased consumption causes the PC decrease.
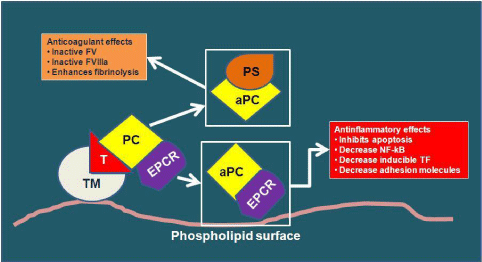
Figure 3: Endogen anticoagulants and their normal functions (TM:
Thrombomodulin; T: Thrombin; PC: Protein C; EPCR: Endothelial Cell
Protein C Receptor; aPC: activated Protein C; PS: Protein S; NF-κB: Nuclear
factor-κB).
Fibrinolysis inhibition
In severe sepsis the fibrinolytic system is compromised. The high production of Plasminogen Activator Inhibitor-1 (PAI-1) acts as a powerful inhibitor of both tissue Plasminogen Activator (t- PA) and urokinase Plasminogen Activator (u-PA). The fibrinolytic system imbalance leads to the significant decreased of Plasmin, whose activity is responsible for dissolving intravascular fibrin clots [23]. The high Thrombin generation and its affinity to bind with the Thrombomodulin, activate the zymogen Thrombin-Activable Fibrinolysis Inhibitor (TAFI) that causes a further decrease in Plasmin generation (Figure 4).
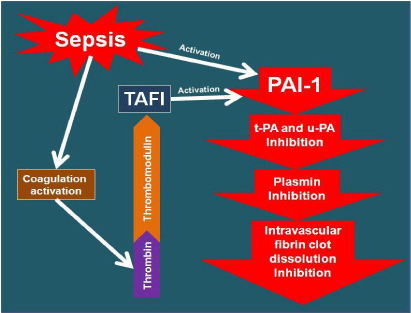
Figure 4: Impairment of the fibrinolytic system in sepsis.
Summarizing, in severe inflammation the impact on the coagulation system can be pivotal, the stressed procoagulant activity increases the susceptibility to thrombosis, Disseminated Intravascular Coagulation (DIC) and MODS, often with an unfavorable outcome (Figures 5 & 6).
Figure : Impact of severe inflammation on the clotting system.
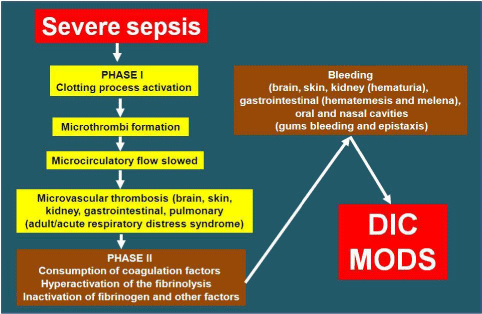
Figure 6: Pathogenetic evolution of severe sepsis in Disseminated
Intravascular Coagulation (DIC) and Multiple Organ Dysfunction Syndrome
(MODS).
Laboratory and inflammation
The inflammatory response in most cases is accompanied with leukocytosis as well as thrombocytosis. The leukogram varies in respect to the normal values for the stress induced by cortisol, as well as for the involvement of the physiological response to epinephrine. Leukocytosis and neutrophilia are the most common responses to inflammation, but a severe inflammation may also manifest an important leukopenia due to bone marrow inhibition or to a massive inflammatory condition. The characteristic morphological changes of neutrophils, such as cytoplasmic vacuoles, cytoplasmic basophilia, Döhle bodies and azurophil granules, underline an inflammatory condition [24]. In non-inflammatory leukocytosis, as well as in patients on appropriate antibiotic therapy, the morphological changes of neutrophils in both cases are not present. The simultaneous effect of glucocorticoids, catecholamines, comorbidities (e.g. cancer), antibiotic therapy, influence the leukogram interpretation significantly, therefore it can be valued by its variability.
Although the Fibrinogen (FBG) is an acute phase protein, it does not allow the correct assessment on inflammatory state because of its limited specificity. The recommended method for its determination is the Clauss assay, whose endpoint is the clot formation. Common source of result, which is not always reliable, is the variability of the reagents; it is appropriate to use single reactive in the context of standardized procedures. Heparin therapy influences its measurement significantly. In fact, at least four hours after heparin administration it is recommended to perform the test, so as not to draw the sample from heparin contaminated-lines [25]. Regarding the data interpretation, the individual variability between patients is another problem [26]. The FBG may be within the normal range when consumed despite overt pathology.
The C-Reactive Protein (CRP) is another acute phase protein that, unlike fibrinogen, in inflammatory focus is distinctly increased. Its increase indicates an inflammatory condition with tissue necrosis since it is expressed on the surface of dead or dying cells in order to activate the complement system through the complex C1Q [27]. The different methods used by laboratories have a good sensitivity and clinical comparability. Since it is an inflammatory marker, the “high sensitivity CRP” assay can predict the risk of myocardial infarction and stroke [28].
Inflammation and main laboratory coagulation tests
It is important to reiterate that the preanalytical error in coagulation has a considerable weight on the analytical data reliability.
The Prothrombin Time (PT) test evaluates the extrinsic and common coagulation pathways efficiency, providing information on factors II, V, VII or X, or on Fibrinogen efficiency. It is frequently required to diagnose acquired bleeding disorder, such as factors Vit. K deficiency, liver disease [29]. The test consists in adding thromboplastin and calcium to citrated blood. The endpoint of the reaction is the clot formation. Depending on the thromboplastin used, the test shows a marked variability. For this reason, the result in patients on oral anticoagulant therapy is expressed as “International Normalized Ratio”, taking into account the thromboplastin “International Sensitivity Index”. The different test sensitivity in recognizing one or more coagulation deficiency factors is due to different sources and concentration of thromboplastin, as well as for phospholipids added [29]. Generally, the test is abnormal when the deficient coagulation factor is < 70%, it is not sensitive to slight coagulation factors deficiency. In DIC consequent to severe inflammation, the PT test is clearly extended. The test in hypercoagulable state, unfractionated heparin bolus, or low molecular weight heparin high doses therapy, does not provide correct information; in lupus anticoagulant associated with thrombotic state interference are also possible [30].
The activated Partial Thromboplastin Time (aPTT) test allows the assessment of any deficiency and inhibition of intrinsic and common coagulation pathways. In the case of an abnormal test, factors II, V, VIII, IX, X, XI or XII, Prekallikrein (PK) and a High Molecular Weight Kininogen (HMWK) deficiency is possible [31]. To monitor unfractionated heparin therapy, the aPTT is the reference test. The increase of FVIII, in conditions that underline an inflammatory state (such as trauma, including surgical trauma, prolonged stress and pregnancy), affect the test. In these cases, the normal test might mask a mild hemophilia A. In PK and HMWK deficiency, the test is abnormal without bleeding and no further diagnostic tests are required.
Fibrinogen (FBG), in addition to the analytical problems, as described above, is synthesized by the liver. The anti-inflammatory and/or anti-thrombolytic therapies affect its values. In massive inflammation and DIC, the fibrinolytic system failure triggers widespread micro thrombi formation and a high consumption of fibrinogen occurs. The consequent formation of high fibrinogen degradation products compromises the fibrinogen function.
Once the fibrinolytic process is triggered, the final product by plasmin degradation on fibrinogen/fibrin-soluble is represented by the D-dimer (D-d) production. Its increase is not only the consequence of a thromboembolic or post-surgical conditions. D-d is increased in neoplastic, cardiac and kidney diseases, immune-mediated hemolytic anemia and secondary coagulopathy, including DIC. The nonspecific inflammatory processes can cause fibrinolysis mediated by neutrophil elastase [32]. The D-d assay, include latex agglutination, immunoturbidimetric and Enzyme-Linked ImmunoSorbent Assay (ELISA) methods. Agglutination tests are not easy to interpret in case of weak positivity; compared to ELISA and immunoturbidimetric methods the agglutination tests have low sensitivity [33]. Elevated D-d levels are associated with a higher risk of overall mortality rate in patients with different diseases [34].
The Antithrombin (AT) is like a negative acute phase protein [35]. In DIC, sepsis and acute thrombotic events, the AT decreases [35]. The chromogenic functional tests are the most reliable for its evaluation [36]. The timing for its measurement evaluation is important; the high consumption in acute thrombosis, or the decrease in case of heparin therapy, do not reflect a real AT deficiency.
Fibrin Degradation Products (FDPs) are generated by fibrinogen, soluble fibrin or cross-linked fibrin degradation. The FDPs test has limited diagnostic reliability in thromboembolic disorders. It only indicates the successful of plasmin activation. The FDPs increase may be caused by a systemic inflammation, hepatic disease and/or kidney defective clearance. The results must be interpreted within the clinical context.
The high degree correlation between inflammation and coagulation highlights how, at the same time, specific tests can, by both systems, be influenced.
In severe sepsis, some coagulation testing may predict the survival rate in the first week and in the first month (Figure 7). In the first week the FBG could be considered as an early prognostic test, however the limitations result from its high variability. As delayed prognostic factors, the PT ratio and AT have a good reliability. If the two tests are assessed by age, hemoglobin, and prognostic score (SAPS II and SOFA score), have a high negative predictive value. Moreover, in the first 24 hours, severe sepsis with constant PT ratio, AT and FBG abnormality tests can lead to an increased mortality indicator over the 28 days [37].
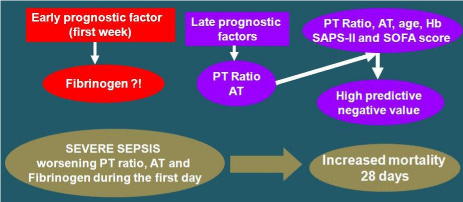
Figure 7: Coagulation testing performed during admission in intensive care
unit that can increase the prediction of survival efficacy in the first week and
in the first month. (PT: Prothrombin Time; AT: Antithrombin).
As part of the cytokines involved in the inflammation/coagulation process, the High Mobility Group Box 1 (HMGB1) protein is taken into clinical consideration. The test is proposed as a possible prognostic marker in DIC and organ failure in septic patients, since HMGB1 is persistently increased in survival septic patients than nonsurvival [38].
Conclusion
In patients with inflammation, the significant involvement of the coagulation system entails the high possibility of thrombotic disease. In diseases whose epiphenomenon is the release of cytokines and/or the natural anticoagulants down-regulation, the interface “inflammation/coagulation” is the most common process of the disease.
Pharmacologically, in septic patients with coagulopathy, an important aspect is to limit the defective action of natural anticoagulants.
Therapeutic strategies have been developed based on the insights into the pathogenetic mechanisms responsible for Thrombin formation and fibrin deposition.
The use of TF inhibitors is still a matter of debate [39]. Also the concentrated AT treatment has not shown significant benefits in reducing mortality in septic patients [40].
Significant results were obtained with recombinant aPC exclusively in patients with severe sepsis, high mortality risk and, consequently, a high prognostic score [41]. Recombinant aPC restores not only the PC anticoagulant, but also has anti-inflammatory action. Even if the trigger conditions are not present, inherited abnormalities of coagulation proteins may be risk factors and promote the pathogenesis of DIC. Although DIC seldom occurs spontaneously, homozygous deficiency of PC or PS in children can cause severe neonatal purpura fulminans, without a specific pathological condition [42].
Since histones in severe septic patients seem to have an important role as critical organ dysfunction mediators, treatment with histones antagonists could be an interesting therapeutic approach in MODS [43].
A pivotal therapeutic strategy could represent the complement inactivation that is released during organ failure, and plays an important role in severe sepsis progression, as well as in clotting interaction [44].
In patients with sepsis and/or significant inflammation, the coagulation monitoring, as well as the correct test interpretation, have an important role because it affects the therapeutic management and the patient’s outcome.
References
- Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. 2002; 30: 294-301.
- Bastarache JA, Ware LB, Bernard GR. The role of the coagulation cascade in the continuum of sepsis and acute lung injury and acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006; 27: 365-376.
- Webster NR. Inflammation and the coagulation system. Br J Anaesth. 2002; 89: 216-220.
- Kaser A, Brandacher G, Steurer W, Kaser S, Offner FA, Zoller H, et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: role in inflammatory thrombocytosis. Blood. 2001; 98: 2720-2725.
- Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004; 25: 5681-5703.
- Esmon CT, Fukudome K, Mather T, Bode W, Regan LM, Stearns-Kurosawa DJ, et al. Inflammation, sepsis, and coagulation. Haematologica. 1999; 84: 254-259.
- Lowe JB. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr Opin Cell Biol. 2003; 15: 531-538.
- GawazM, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C. et al. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis. 2000; 148: 75-85.
- Burstein SA. Cytokines, platelet production and hemostasis. Platelets. 1997; 8: 93-104.
- Saluk-Juszczak J, Wachowicz B, Kaca W. Stimulatory effects of endotoxin on the platelet secretory process. Microbios. 1999; 99: 45-53.
- Selak MA, Chignard M, Smith JB. Cathepsin G is a strong platelet agonist released by neutrophils. Biochem J. 1988; 251: 293-299.
- Dugina TN, Kiseleva EV, Chistov IV, Umarova BA, Strukova SM. Receptors of the PAR family as a link between blood coagulation and inflammation. Biochemistry (Mosc). 2002; 67: 65-74.
- Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, et al. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001; 154: 485-490.
- Esmon CT. The impact of the inflammatory response on coagulation. Thromb Res. 2004; 114: 321-327.
- Boos CJ, Goon PK, Lip GY. The endothelium, inflammation, and coagulation in sepsis. Clin Pharmacol Ther. 2006; 79: 20-22.
- van Hinsbergh VW. Endothelium--role in regulation of coagulation and inflammation. Semin Immunopathol. 2012; 34: 93-106.
- Riewald M, Ruf W. Orchestration of coagulation protease signaling by tissue factor. Trends Cardiovasc Med. 2002; 12: 149-154.
- Danese S, de la Motte C, Rivera Reyes BM, Sans M, Levine AD, Fiocchi C. T cells trigger CD40-dependent platelet activation and granular RANTES release: A novel pathway for immune response amplification. The Journal of Immunology. 2004; 172: 2011–2015.
- Danese S, Fiocchi C. Platelet activation and the CD40/CD40 ligand pathway: mechanisms and implications for human disease. Crit Rev Immunol. 2005; 25: 103-121.
- Viles-Gonzalez JF, Fuster V, Badimon JJ. Links between inflammation and thrombogenicity in atherosclerosis. Curr Mol Med. 2006; 6: 489-499.
- Kälsch T, Nguyen XD, Elmas E, Grebert N, Süselbeck T, Klüter H, et al. Coagulation activation and expression of CD40 ligand on platelets upon in vitro lipopolysaccharide-challenge in patients with unstable angina. Int J Cardiol. 2006; 111: 217-223.
- Esmon CT. Coagulation inhibitors in inflammation. Biochem Soc Trans. 2005; 33: 401-405.
- Nesheim M. Thrombin and fibrinolysis. Chest. 2003; 124: 33-39.
- Stock W, Hoffman R. White blood cells 1: non-malignant disorders. Lancet. 2000; 355: 1351-1357.
- Mackie IJ, Kitchen S, Machin SJ, Lowe GD. Haemostasis and Thrombosis Task Force of the British Committee for Standards in Haematology. Guidelines on fibrinogen assays. Br J Haematol. 2003; 121: 396-404.
- Machin SJ, Mackie IJ. Routine measurement of fibrinogen concentration. BMJ. 1993; 307: 882-883.
- Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999; 7: 169-177.
- Ridker PM. Cardiology Patient Page. C-reactive protein: a simple test to help predict risk of heart attack and stroke. Circulation. 2003; 108: 81-85.
- Chee YL, Greaves M. Role of coagulation testing in predicting bleeding risk. Hematol J. 2003; 4: 373-378.
- Greaves M. Assessment of haemostasis. Vox Sang. 2004; 87: 47-50.
- Taylor LJ. Laboratory management of the bleeding patient. Clin Lab Sci. 2003; 16: 111-114.
- Wada T, Gando S, Yanagida Y, Uegaki S, Kubota N. Elastase-mediated fibrinolysis in disseminated intravascular coagulation. 22nd International Congress on Thrombosis. 2012.
- Harper P, Marson C, Grimmer A, Monahan K, Humm G, Baker B. The rapid whole blood agglutination d-dimer assay has poor sensitivity for use as an exclusion test in suspected deep vein thrombosis. N Z Med J. 2001; 114: 61-64.
- Di Castelnuovo A, de Curtis A, Costanzo S, Persichillo M, Olivieri M, Zito F, et al. Association of D-dimer levels with all-cause mortality in a healthy adult population: findings from the MOLI-SANI study. Haematologica. 2013; 98: 1476-1480.
- Niessen RW, Lamping RJ, Jansen PM, Prins MH, Peters M, Taylor FB, et al. Antithrombin acts as a negative acute phase protein as established with studies on HepG2 cells and in baboons. Thromb Haemost. 1997; 78: 1088-1092.
- Kottke-Marchant K, Duncan A. Antithrombin deficiency: issues in laboratory diagnosis. Arch Pathol Lab Med. 2002; 126: 1326-1336.
- Lissalde-Lavigne G, Combescure C, Muller L, Bengler C, Raillard A, Lefrant JY, et al. Simple coagulation tests improve survival prediction in patients with septic shock. J Thromb Haemost. 2008; 6: 645-653.
- Hatada T, Wada H, Nobori T, Okabayashi K, Maruyama K, Abe Y, et al. Plasma concentrations and importance of High Mobility Group Box protein in the prognosis of organ failure in patients with disseminated intravascular coagulation. Thromb Haemost. 2005; 94: 975-979.
- Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, et al. OPTIMIST Trial Study Group. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003; 290: 238-247.
- Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, et al. Opal SM; for the KyberSept Trial Study Group. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001; 286: 1869-1878.
- Levi M, Schultz M, van der Poll T. Disseminated intravascular coagulation in infectious disease. Semin Thromb Hemost. 2010; 36: 367-377.
- Koster T, Rosendaal FR, Briet E, van der Meer FJ, Colly LP, Trienekens PH, et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study) Blood. 1995; 85: 2756-2761.
- Chaput C, Zychlinsky A. Sepsis: the dark side of histones. Nat Med. 2009; 15: 1245-1246.
- Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of E coli sepsis. Blood. 2010; 116: 1002-1101.