
Special Article - Thalassemia
J Blood Disord. 2020; 7(1): 1057.
Assessment of Hematological Characteristics among β -Trait & Hb-E Trait Individuals in Bangladesh
Aziz A1,2*, Bilkis A1 and Khan WA2
1Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Bangladesh
2Department of Biochemistry and Molecular Biology, Dhaka Shishu (Children) Hospital, Bangladesh
*Corresponding author: Aziz A, Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Department of Biochemistry and Molecular Biology, Dhaka Shishu (Children) Hospital, Bangladesh
Received: January 11, 2020; Accepted: February 19, 2020; Published: February 26, 2019
Abstract
Thalassemia are an outfit of autosomal recessive disorders caused by reduction or absent production of one or more of the globin chains. About 3% people are carriers of β-thalassemia and 4% are E- β-thalassemia in Bangladesh. The study was undertaken to determine hematological characteristics among β-thalassemia trait & Hb E trait individual & the effect of different mutations with hematologic parameters. A total 280 blood samples were included for this study and the samples were analyzed by Siemens Advia 2000i Hematology Analyzer and automated capillarys-2 flex piercing system. The samples were analyzed in the Department of Biochemistry and Molecular Biology of Dhaka Shishu (Children) Hospital, Dhaka, Bangladesh. After extraction of DNA, beta globin gene mutations were detected by DNA sequencing method & data were analyzed by SPSS, Version 20, IBM Cor. The study found a statistically significant comparison of mean Hb A2 (%) between common mutations & rare mutations of beta trait population (p‹0.05). In Hb E trait population, Hb A2 was (3.44±0.6%) & (p‹0.001) and Hb E was (26.5±2.54%) & p value was 0.000. Hb E trait population showed comparatively higher values of HCT, MCV and MCH than beta thalassemia trait groups (p‹0.05). No significant difference was observed in RDW, MCHC & Hb (g/dl) between the two groups (p›0.05). The study will be helpful in improving population screening for identification the carriers of β-thalassemia in Bangladesh.
Keywords: Autosomal; β-thalassemia; Mutations; Hematological characteristics
Introduction
Thalassemia is a conditions in which there are reduced rate of synthesis of one or more of the globin chains leading to an imbalanced synthesis of globin chains and production of defective hemoglobin [1]. The incidence for this disease is high in the tropical and subtropical areas including Southeast Asia [2]. The carrier rate of β-thalassemia is 3.0% and Hb-E/β- thalassemia is 4.0% and affected birth per thousand of β-thalassemia and Hb-E/ β-thalassemia is 0.106 & 3.000 respectively in Bangladesh [3,4]. About 10% of the world’s thalassemia major kiddies are born in India [5]. The carrier circumstance varies from country to country. Hb E is the most common abnormal found in Southeast Asia including Bangladesh, India, Cambodia, Laos, Myanmar and Thailand [6]. Haemoglobin E allele, point mutation (G > A) in codon 26 of β - globin gene that can induce alternative splicing and thus result in decreased β -globin E chains [7]. Hb E/β-thalassemia results from co-inheritance of a β-thalassemia allele from one parent and Hb E from other parent [8]. HbE/ β thalassemia causes a surprisingly variable anaemia, ranging from nearly asymptomatic states to severe anemia [7]. The average beta thalassemia carrier prevalence is 5% in Pakistan, 3.1% in Tunisia & 3.3% in India [9-11]. In Arab countries, the prevalence of beta thalassemia trait was 3% in Oman, 8.7% in United Arab Emirates, 2.9% in Bahrain [12-14]. Almost every possible defect affecting gene expression at transcription or post-transcriptional level including translation had been identified in β thalassemia [15]. These genetic defects lead to a variable reduction in β globin output that ranges from a minimal deficit (mild β+ thalassemia alleles) to complete absence (β° thalassemia). β-thalassemia can be divided into three main types depending on clinical phenotypes: thalassemia major, thalassemia trait and thalassemia intermediate [16]. β-thalassemia major is a severe form that requires blood transfusions from infancy for survival. This clinical phenotypic variability of β-thalassemia occurs due to the mutation in three exons and two intervening sequences 5´ UTR and the 3´ UTR of β-globin (HBB) gene [17,18]. Mutation in exons and intervening sequences that may produce nonfunctional beta globin protein or β0 allele [19]. Approximately 600 mutations have been found in the β-globin (HBB) gene of which more than 200 are associated with β-thalassemia phenotype [20]. The distribution and frequency of different mutations pertaining thalassemia vary from population to population. One reason of prevalence of thalassemia are intermarriage between different ethnic groups, lack of awareness for blood test before marriage [21]. Many countries have started control programme to prevent the births of thalassemic children as prevention is more cost effective than treating thalassemic patients [22]. The study was undertaken to determine hematological characteristics & the effect of different mutations on hematologic parameters in beta-thalassemia & Hb E trait individual to improve population-screening program in Bangladesh.
Materials and Methods
The study was conducted in dept. of biochemistry and molecular biology, Dhaka Shishu (Children) Hospital, Dhaka, Bangladesh.
Study subject
The study included 280 individuals, all of whom were selected by testing Hb capillary electrophoresis. Estimation carried out by fully automated capillarys-2 flex piercing system. Individual who had Hb A2 level › 3.5% were diagnosed as beta thalassemia carrier and Hb E carrier when E band constituted 22-30%. Hb capillary electrophoresis data of each sample was documented for analysis.
Sample collection
About 5.0 mL of venous blood was drawn & taken in an EDTA coated tube from each individual following all aseptic precautions with the help of a trained person. Complete blood counts and red blood cells were estimated by automated Hematology analyzer. These hematological data of each sample were recorded in order to performing analysis. Blood samples were stored at -20o c until molecular analysis.
Extraction of genomic DNA
Genomic DNA was extracted from whole blood sample using the Invitrogen Kit (Invitrogen, USA) by manufacturing process. The extracted DNA was stored at –20 °C for further use.
Polymerase chain reaction amplification and sequencing
Firstly, the genomic DNA was amplified by Exon I, II and III specific primer and the polymerase chain reaction (PCR) method. These PCR products were run in 2% agarose gel and the bands were visualized using the trans UV illuminator. Sequencing reaction was performed by Big Dye terminator cycle sequencing kit (Applied Bio systems, USA). Analysis was done by automated capillary electrophoresis in the 310 genetic analyzer (Applied Bio systems, USA).The sequences that are obtained from the capillary electrophoresis were aligned using the Reference Sequence of the β globin gene [NCBI Ref Seq entry of HBB (NG_000007.3)] in the Seqscape sequence alignment version 2.5 (Applied Bio systems, USA). Then clinical significance and genetic variants were observed by HbVar database (http://globin.bx.psu.edu/hbvar) of haemoglobin variants and thalassemia mutations.
Statistical analysis
Data were entered and analyzed, using SPSS, Version 20, IBM Cor. Data were expressed as (mean±SD) and number (percentage). The (mean±SD) of each hematologic parameter were calculated & t-tests & chi square test were performed where p‹0.05 was considered significant.
Results
Among the study subject, 70% population had beta trait & 30% had Hb E trait. 36.43% were male and 33.57% were female among the beta trait population & 15.36% male and 14.64% female were Hb E trait individual (Tables 1 & 2).
![]()
Trait
Gender
Frequency (N= 280)
Percentage (%)
Beta trait
Male
102
36.43 %
Female
94
33.57 %
Total
196
70.00 %
Hb E trait
Male
43
15.36 %
Female
41
14.64 %
Total
84
30.00 %
Table 1: Demographic characteristics of study subjects.
![]()
Parameters
Beta trait
(mean ± SD)
Hb E trait
(mean ± SD)
P value
Hb (g/dl)
11.07 ± 1.34
12.26 ± 0.02
P>0.05
HCT (%)
26.5 ± 2.8
37.06 ± 1.7
P<0.05
RDW (%)
16.35 ± 2.143
14.17 ± 0.643
P>0.05
MCH (pg)
20.71 ± 2.012
29.05 ± 1.125
P<0.05
MCV (fl)
63.81 ± 5.324
77.20 ± 4.217
P<0.05
MCHC (g/dl)
33.55 ± 1.586
33.07 ± 2.49
P>0.05
Table 2: Comparisons of hematological characteristics between beta trait & Hb E trait population.
The Hb E trait population group showed comparatively higher values for hematological indices (HCT, MCV and MCH) than beta thalassemia trait groups (p‹0.05). No significant difference was observed in RDW, MCHC & Hb (g/dl) between two groups (p>0.05) (Figure 1 & Table 3).
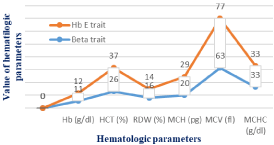
Figure 1: Comparisons of hematological parameters between Beta trait & Hb
E trait population.
![]()
Mutations
Frequency
(N)
Percentage
(%)
A2 level (%)
(mean ± SD)
IVS 1-5 (G>C)
138
70.40
5.08 ± 0.55
CD 15 (G>A)
8
4.08
5.56 ± 0.78
Fr 41-42 (-CTTT)
14
7.1
5.92 ± 0.72
Fr 8/9 (+G)
10
5.1
5.59 ± 0.51
CD 30 (G>C)
11
5.6
5.51 ± 0.41
IVS 1-130 (G>C)
8
4.08
5.22 ± 0.41
Fr 16 (+G)
2
1.0
6.02 ± 0.8
CD 15 (-T)
3
1.5
6.06 ± 0.40
-90 (C>T)
2
1.0
6.67 ± 0.44
Table 3: Frequency of mutations among beta trait population.
Among total nine beta trait mutations, IVS 1-5 (G›C) was the commonest mutation for 70.40% of cases where mean Hb A2% value was (5.08±0.5). Another common mutation were Fr 41-42 (-CTTT), Fr 8/9 (+G), CD 30 (G›C) found in found in 7.1%, 5.1% & 5.6% of cases respectively & mean Hb A2 was (5.92±0.72), (5.59±0.51) & (5.51±0.41) respectively. For IVS 1-130 (G›C) (4.8%), CD 15 (G›A) (4.8%) where mean Hb A2% was (5.22±0.41) & (5.22±0.41) respectively. Several rare mutations, -90 (C›T) (1.0%), CD 15 (-T) (1.5%), Fr 16 (+G) (1.0%) were also found where mean Hb A2% was (6.02±0.8), (6.06±0.40) & (6.67±0.44) respectively (Table 4).
![]()
Mean A2%
P value
Common mutations
Rare mutations
< 0.05
(5.48 ± 0.56)
(6.25 ± 0.55)
Table 4: Comparison of mean A2% between common mutations & rare mutations among beta trait population.
The study found a statistically significant difference on A2 (%) and beta trait mutation. Rare beta trait mutations had higher Hb A2 (%) than other common beta trait mutations where and the p value was ‹0.05. Mean Hb A2% for common mutations was (5.48±0.56) & for rare mutations was (6.25±0.55) (Table 5).
![]()
Hb E trait mutation
Electrophoretic characterization
Cd 26 (G>A)
Value
Minimum
Maximum
Mean ± SD
Chi square value
df
P value
Hb A2 (%)
2
6
3.44 ± 0.6
168.000
12
<0.001
Hb E (%)
22
32.95
26.5 ± 2.54
146.213
8
0.000
Table 5: Measurement of Hb A 2% & Hb E% value in capillary HPLC for Hb E trait population.
The study showed that, the mean capillary electrophoresis value of Hb A2 was (3.44±0.6%) where the p value was ‹0.001. The mean value of Hb E was (26.5±2.54%) where & p value was 0.000.
Discussion
Beta thalassemia is a congenital disorder that is prevalent in certain parts of the world including Bangladesh. As a result of geographical position and global population movement, β-thalassemia had become a common hemoglobin genetic disorder in Bangladesh [23,24]. Gene flow from Indian sub-continent to Bangladesh may be one of the motives of prevalence of this disease in Bangladesh [4]. Information regarding mutation spectrum of thalassemia patient is yet scarce in our country at national level. Moreover, investigation of mutation information is not a usual practice in our country. Thalassemia patients are initially suspected emerging on common hematological parameter screening followed by hemoglobin electrophoresis. This study had identified nine beta globin gene mutations & IVS 1-5 (G›C) was the commonest mutation for 70.40%. This mutation reported as the most common mutation in Indian population [25] & in UAE (55%) and Oman (62%) [11,12]. In this study, among total nine beta trait mutations, IVS 1-5 (G›C) had mean Hb A2 value of (5.08±0.5). Another followed mutation Fr 41-42 (-CTTT), Fr 8/9 (+G), CD 30 (G›C) found in found in 7.1%, 5.1% & 5.6% of cases respectively & mean Hb A2 was (5.92±0.72), (5.59±0.51) & (5.51±0.41) respectively. For IVS 1-130 (G›C) (4.8%) & CD 15 (G›A) (4.8%) where mean Hb A2 was (5.22±0.41) & (5.22±0.41) respectively. Several rare mutations also found were, -90 (C›T) (1.0%), CD 15 (-T) (1.5%), Fr 16 (+G) (1.0%) where mean Hb A2 was (6.02±0.8), (6.06±0.40) & (6.67±0.44) respectively. The statistically significant of mean A2 (%) between common mutations & rare mutations among beta trait population where rare beta trait mutations had higher Hb A2(%) than other common beta trait mutations (p‹0.05). Hb-E (Codon 26 G>A) was the second most common mutation in 84 out of 280 cases (30%) in this study. The study found that mean Hb A2% (3.44±0.6%); (p‹0.001), & mean Hb E% (26.5±2.54%); (p‹0.000) in Hb E trait population.
In this study, the mean RBC count in male & female were (5.91±0.61) m/μl & (5.34±0.46) m/μl respectively. The HCT was (33.57±3.3)% for men and (32.9±2.19)% for women respectively. MCH was (20.71±2.012) pg with minimum value of 18 and maximum value of 28. The mean value of MCV per fL of blood was 63.81 and standard deviation was 5.324 where reference value for adult was (92±9) fL. Among the beta trait population, MCHC value was (33.55±1.586)g/dl, which was normal as compared to reference value. RDW was (16.35±2.143)% where reference value for adult was (12.8±1.2)%. The (mean±SD) of Hb & HCT in Hb E trait male were (13.02±1.56) & (38.81±5.15). In female, the mean±SD of Hb & HCT were & (11.5±0.88) g/dl & (34.51±1.03)% respectively. In Hb E trait population, both MCV (77.20±4.217) fL & MCH (26.05±1.125) pg were slightly reduced where MCV & MCH was markedly reduced in beta trait population. In Hb E trait population, mean RDW (14.17±0.643)% was higher than reference value but mean MCHC (33.07±2.49) g/dl was normal.
The Hb E trait population group showed comparatively higher values for hematological indices (MCV, HCT and MCH) than beta thalassemia trait groups (p‹0.05). Inherited disorders of hemoglobin synthesis cause morbidity and mortality worldwide & place a large burden to the patients and their families [26]. Thalassemia are generally not curable but can be prevented by population screening, genetic counseling and prenatal diagnosis.
Acknowledgement
We thank Bilquis Banu (Professor, Department of Clinical pathology, Dhaka Shishu (Children) Hospital, Bangladesh) for encouragement and inspiration to complete the research work.
References
- Marengo-Rowe AJ. The thalassemias and related disorders. Proc (Bayl Univ Med Cent). 2007; 20: 27-31.
- Rahman MJ, Rahman MH. Prevention & control strategy of thalassemia in Bangladesh. The Orion Med J. 2003; 16: 121-122.
- Sengupta M. Thalassemia among the tribal communities of India. The Internet Journal of Biological Anthropology. 2007; 1: 2.
- Aziz A, Ahmed W, Sarwardi G, Ara Naznin R, Rehena J, Ferdoushi A. Molecular Analysis of Hb-E and Beta-Thalassemia Major Patients among Bangladeshi Population. Austin J Biotechnol Bioeng. 2017; 4: 1085.
- Chen W, Zhang X, Shang X, Cai R, Li L, Zhou T, et al. The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity. BMC Med Genet. 2010; 11: 31.
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres ClinHaematol. 1998; 11: 1-51.
- Orkin SH, Kazazian HH Jr, Antonarakis SE, Ostrer H, Goff SC, Sexton JP. Abnormal RNA processing due to the exon mutation of beta E-globin gene. Nature. 1982; 300: 768-769.
- Huisman TH, Carver MF. The beta- and delta-thalassemia repository (Ninth Edition; Part I). Hemoglobin. 1998; 22: 169-195.
- Ambekar SS, Phadke MA, Mokashi GD, Bankar MP, Khedkar VA, Venkat V, et al. Hemoglobinopathies in Western MaharashtaIndian Paediatrics. 2001; 38: 530-534.
- Mseddi S, Gargouri J, LabiadhZ, Kassis MA, Elloumi M, Ghali L, et al. Prevalence of Haemoglobin abnormalities in Kibli. Rev Epidemiol Sante Publique. 1999; 47: 29-30.
- Al-Riyami A, EbrahimGJ. Genetic Blood disorders Survey in Sultanate of Oman. J Trop Pediatric. 2003; 49: 1-20.
- Miller CJ, Dunn EV, Beg B, Abdouni SF. A haematological survey of preschool children of United Arab Emirates. Saudi Med J. 2003; 24: 609-613.
- Al-Arrayed S, Hafadh N, Amin S, Al-Mukhareq H, SanadH. Student screening for inherited blood disorders in Bahrain. East Mediterr Health Journal. 2003; 9: 344-345.
- Balgir RS. The genetic burden of hemoglobinopathies with special reference to community health in India and the challenges ahead. Indian Journal of Hematology and Blood Transfusion. 2002; 20: 2-7.
- Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013; 3: a011700.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010; 5: 11.
- Kulkarni GD, Kulkarni SS, Kadakol GS, Kulkarni BB, Kyamangoudar PH, Lakkakula BV, et al. Molecular basis of β-thalassemia in Karnataka, India. Genet Test Mol Biomarkers. 2012; 16: 138-141.
- Vetter B, Neu-Yilik G, Kohne E, Arnold R, Sinha P, Gaedicke G, et al. Dominant beta-thalassaemia: a highly unstable haemoglobin is caused by a novel 6 bp deletion of the beta-globin gene. Br J Haematol. 2000; 108: 176-181.
- Al-Arrayed S, Hafadh N, Amin S, Al-Mukhareq H, Sanad H. Student screening for inherited blood disorders in Bahrain. East Mediterr Health Journal. 2003; 9: 344-345.
- Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004; 124: 264-274.
- Khattak MF, Saleem M. Prevalence of heterozygous in Northern areas of Pakistan. J Pak Med Assoc. 1992; 42: 32-34.
- Galanello R, Eleftheriou A, Old J, Angastniotis M, Synodinos JT, Jauniaux E, et al. Prevention of Thalassemias and other Haemoglobin disorders. Thalassemia International Federation Publications, Nicosia, Cyprus. 2013.
- Khan WA, Banu B, Sadiya S, Sarwardi G. Spectrum of types of thalassemias and hemoglobinopathies: study in a tertiary level children hospital in Bangladesh. Thalassemia Reports. 2017; 7: 6354.
- Abdul A, Das SA, WA Khan, Sadiya S, Banu B, Sarwardi G, et al. A Novel β-Thalassemia Insertion/Frameshift Mutation between Codons 77/78 (p.Leu78Profs*13 or HBB: c.235_236insC) Observed in a Family in Bangladesh, Hemoglobin. 2017; 41: 311-313.
- Panigrahi I, Marwaha R. Mutational spectrum of thalassemias in India. Indian journal of human genetics. 2007; 13: 36-37.
- Bilgen T, Clark OA, Ozturk Z, Akif Yesilipek M, Keser I. Two novel mutations in the 3’ untranslated region of the beta-globin gene that are associated with the mild phenotype of beta thalassemia. Int J Lab Hematol. 2013; 35: 26-30.