
Research Article
J Blood Disord. 2021; 8(1): 1063.
Molecular Investigation of Iranian Patients Suspected to Hereditary Spherocytosis
Shahab-Movahed Z1, Majd A1, Torbati ES2 and Zeinali S3,4*
1Department of Cellular and Molecular Biology, Faculty of Biological Sciences, Islamic Azad University, North Tehran Branch, Tehran, Iran
2Department of Genetic, Faculty of Science, North Tehran Branch of Islamic Azad University, Tehran, Iran
3Department of Medical Molecular, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran
4Kowsar Human Genetic Research Center, Tehran, Iran
*Corresponding author: Sirous Zeinali, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran
Received: May 31, 2021; Accepted: June 18, 2021; Published: June 25, 2021
Abstract
Introduction: Hereditary spherocytosis is a heterogeneous disorder with mild to moderate anemia. The aim of this study was to evaluate the inherited spherocytosis gene mutations in patients with RBC cytoplasmic disorders in Iranian population.
Materials and Methods: In this study, Whole Exome Sequencing (WES) was performed for patients suspected to hereditary spherocytosis and their relatives.
Results: Sequence analysis of the probands and their parents identified variations in ANK1 gene (NM_001142446.1:c.127-2A>G), SPTB (c. 14delC, p.Thr5LysfsTer41), SPTA1 (c.466C>T), SLC4A1 (c.2494C>T) and SLC25A38 gene (c.683G>T, NP_060345.2:p.Gly228Val that could be related to the patients clinical manifestation.
Conclusion: Findings are in line with the appropriate diagnostic yield of WES in determining the causative variant especially in those disorders that many genes are involved like anemia. This is the first report of a cohort of Iranian patients with anemia suspected to that were investigated using WES technology. Further studies are needed to investigate the distribution of gene mutations in patients with RBC membrane disorders in Iran
Keywords: Hereditary spherocytosis; Whole-exome sequencing; Hemolytic anemia; Erythrocyte membrane protein; ANK1
Abbreviations
HS: Hereditary Spherocytosis; DRTA: Hereditary Distal Renal Tubular Acidosis; AE1: Anion Exchanger 1; WES: Whole Exome Sequencing; ACMG: American College of Medical Genetics; ANK1: Ankyrin 1; EPB42: Erythrocyte MEMBRANE PROTEIN BAND 4.2; HS: Hereditary Spherocytosis; MCH: Mean Corpuscular Hemoglobin; MCHC: Mean Corpuscular Hemoglobin Concentration; MCV: Mean Corpuscular Volume; PCR: Polymerase Chain Reaction; RBCs: Red Blood Cells; SLC4A1: Solute Carrier Family 4 Member 1; SPTA1: Spectrin a Erythrocytic 1; SPTB: Spectrin β;Variants of Unknown Significance (VUS)
Introduction
Red cell membrane disorders that affect its membrane stability results in irregular red cell shape and disorders including Hereditary Spherocytosis (HS), Hereditary Elliptocytosis (HE), Hereditary Pyropoikilocytosis (HPP) and Hereditary Stomatocytosis (HSt) [1,2]. Hereditary spherocytosis (HS) refers to a common heterogeneous group of congenital hemolytic anemia due to red blood cell structural proteins disorder resulting in spherical-shaped erythrocytes on the peripheral blood smear [1] with wide clinical heterogeneity ranging from asymptomatic cases to severe hemolytic anemia [3] besides reticulocytosis, jaundice, splenomegaly, increased indirect bilirubin and red blood cell osmotic fragility. The worldwide incidence is about one in 2,000 to 5,000 Caucasians [4] and few cases were reported in the black population [2,5] with dominant inheritance in most cases (75%) [6]. Five genes correlated with the pathogenesis of HS so far including Ankyrin (ANK1) (most common (7)), a-spectrin (SPTA1), β-spectrin (SPTB), band 3 (SLC4A1) and protein 4.2 (EPB42) that interplay between the erythrocyte membrane and the lipid bilayer [8]. Defects in protein product of these genes, result in loss of vertical junction between cell membrane and lipid bilayer [1]. High-throughput Next Generation Sequencing (NGS) technology has promoted rapid and cost-effective genetic diagnosis of various genes, especially when routine laboratory testing fails [9,10]. Here, we present clinical and molecular genetics of Iranian cases with HS investigated with whole exome analysis to find the causative mutations to provide prenatal diagnosis in the future pregnancies.
Materials and Methods
Patients and study design
Patients with anemia and primary clinical diagnosis of Hereditary Spherocytosis (HS) were referred from the Blood disorder clinic of Ali-Asghar Children’s Hospital, Iran University of Medical Sciences, Tehran, Iran to Dr. Zeinali’s Medical Genetics Lab, from September 2017 to March 2018 for genetic counseling and genetic diagnosis. Each patient was reviewed to determine clinical features, laboratory data, disease history (age at diagnosis, jaundice, transfusion history and age at first transfusion and family history). Families with clinical evidence of congenital hemolytic anemia suspected to HS were recruited to the study. After genetic counseling, drawing family pedigree, recording the clinical characteristic and obtaining informed consent, peripheral blood sample (10 ml) were taken from patients and other family members on tubes containing EDTA. Hematologic tests including Complete Blood Count and Analysis (CBC), reticulocyte count, NaCl osmotic fragility test (on fresh and incubated blood), red blood cell distribution width (RDW), peripheral blood smear examination, Direct Antiglobulin Test (DAT) and hemoglobin electrophoresis were performed at Ali-Asghar Children’s Hospital laboratory. Genomic DNA was extracted using salting out Proteinase K method (Kawsar Biotech Co., Tehran, Iran) following the manufacturer’s instruction. Quantity and quality of isolated DNA were measured by Nanodrop spectrophotometer (ThermoScientific®, Waltham, USA) and gel electrophoresed on 1% agarose, respectively. This study was approved by the ethics committee of Kawsar Human Genetics Research Center (KHGRC).
Whole genome sequencing was performed using Illumina’s HiSeq X-ten machines (deCODE Genetics, Reykjavik, Iceland) (Supple S1). More than 95% of the targeted sequences being covered adequately for high-confidence variant calling (>30 X coverage). Rare variants with minor allele frequency less than 1% in the population were included in final analysis. We considered coding and splicing variants with high impact (stop, frameshift, and splice site) and moderate impact (missense, in frame and splice region) on protein function and focused on Single-Nucleotide Polymorphisms (SNPs) and small indels (< 20 base pairs) and prioritized the analysis on mutations in phenotypically relevant genes, i.e. genes with a known link to Mendelian disorders according to Online Mendelian Inheritance in Men (OMIM) (http://www.omim.org) that fit the phenotypes of the index and the inspected inheritance model (especially those relevant to HS including ANK1 (HS type 1, OMIM #182900), SPTB (HS type 2, OMIM #616649), SPTA1 (HS type 3, OMIM #270970), SLC4A1 (HS type 4, OMIM #612653), and EPB42 (HS type 5, OMIM #612690) genes). Vari¬ants in candidate genes that were suspected to be involved in the disease pathogenesis (Variants of unknown significance (VUS), Likely Pathogenic and Pathogenic) were selected for Sanger sequencing in the proband, her/his parents and other family members, if present. Specific forward and reverse primers according to the identified variants were designed with NCBI Primer- Blast Tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast) and synthesized commercially by Metabion Co., Germany. A list of the designed primers and amplification reaction were provided in Supple S2 and Table S1.
Results
Twenty-five samples (9 affected cases and 16 healthy family members from 5 consecutive families (consanguineous marriage in 2 families) were enrolled to the study by anemia and primary clinical diagnosis of Hereditary Spherocytosis (HS). Clinical characteristics and laboratory data in probands at the time of first visit are summarized in Table S1. Following variant filtering of WES results in the probands and their parents, two pathogenic/likely pathogenic variants in ANK1 and SPTB genes were identified in two separate families and confirmed by segregation analysis within the family by direct Sanger sequencing (Table 1, Figure 1 and Figure 2). Furthermore, we identified some candidate variants of uncertain significance in other families that underwent further segregation study within the family to confirm their presence and pathogenicity, as follows.
![]()
Family ID
Gene
Position (hg38) HGVSc/HGVSp
Variant type (Exon No/ Total exon No)
ACMG classificationIinheritance
363821
ANKl
Chr8: 41758139
NM_001142446.l:c. 127-2A>GHetero, splice_acceptor_variant, lntron 1/42
Pathogenic variant, AD
364002
SPTB
Chr14: 64823080
NM_001024858.2:c.14 dclC
NP_001020029.1:p.Thr5LysfsTcr41Hetero, Framesh ift_variant Exon 2/36
Pathogenic variant, de novo
3364803
SLC4Al
chr17:44251320
NM_000342.3:c.2494C>T NP_000333.1:p.Arg832CysHomo, Missense variant Exon19/20
VUS, AR
SPTAl
chr1:158681592
NM _003126.2:c.466C>T
NP_003117.2:p.Argl 56TrpHetero, Missense variant, Exon4/52
VUS, AD
366364
HBB
Chr11: 5226930 NM_00051 804:c.92G>C
NP_000509.1:p.Arg3lThrmissence variant 1/3
Liekly pathogenic AD
EPB41
chr1: 29033108
NM_001166005.1:c.1228G>T
NP_001159477.1:p.Asp4 10TyrHet (affected father) misscnce_variant, 9/21
vus
PIEZO1
chrl 6:88732469
NM_001142864.2:c.2857C>T NP_001136336.2:p.Arg953CysHet (affected father) missence variant 21/51
vus
KIF22
chr16:29803561
NM-007317.2:c.1563-1564delAG
NP_015556. 1:p.Ala523LysfsTer3IHet (affected son frameshift_variant
HIGH impact
10/14Liekly pathogenic AD
NOTCH3
chr1 9:15181721
NM 000435.2:c.2647C>T NP_000426.2:p.Arg883TerHet {affected son) stop_gained_vaiiant, HIGH impact
17/33Liekly pathogenic AD
318635
SLC25A38
Chr3:39394467
NM_017875.2:c.683G>T
NP_060345.2 p.Gly228ValHomo, rnissence_variant, (6/7)
Likely pathogenic, AR
Het: Heterozygous; AD: Autosomal Dominant; VUS: Variant of Uncertain Significance.
Table 1: Assessing the pathogenicity of mutations identified in 5 Iranian families, using bioinformatics tools and ACMG Standards.
In family 1 (family 363821), the proband was a 13 YO girl with neonatal presentation of jaundice and pallor. Clinical feature and the pedigree were shown in Table S1 and Figure 1 respectively. Similar presentation and diagnosis were performed for her mother who underwent splenectomy and cholecystectomy. Sequencing the affected child, her affected mother and healthy father, maternal aunt and grandmother revealed a heterozygote pathogenic splicing variant in ANK1 gene (NM_001142446.1:c.127-2A>G) in the affected cases (mother and daughter) that were confirmed by Sanger sequencing (Table 2). The variant is located in a conserved position with GERP score = 4.37 and in silico analysis with Human Splicing Finder (HSF) predicted it as most probably affecting splicing.
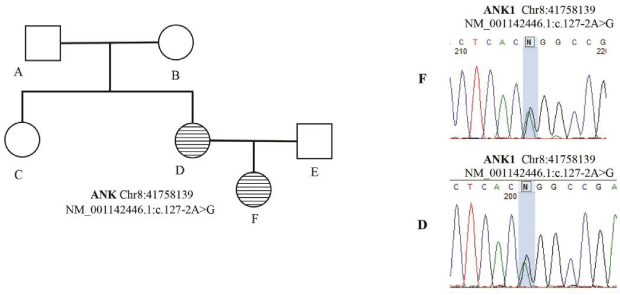
Figure 1: Genetic genealogy of family 363821.
In family 2 (family364002), the proband was an 18 YO boy with neonatal presentation of jaundice, pallor and hyperbilirubinemia. Clinical feature and the pedigree were shown in Table S1 and Figure 2 respectively. No history of hemolytic anemia was noted in his parents. He underwent splenectomy and cholecystectomy due to severe anemia at age 6 YO and clinical and laboratory presentation of the anemia was ameliorated after surgery. Sequencing the affected child, her healthy parents and, sibling and grandmothers revealed a heterozygote pathogenic frameshift de novo variant in SPTB gene (c. 14delC, p.Thr5LysfsTer41) in the affected case that was absent in the other healthy family member (confirmed by Sanger sequencing) (Figure 2). The variant is located in a conserved position with GERP score = 5.05.
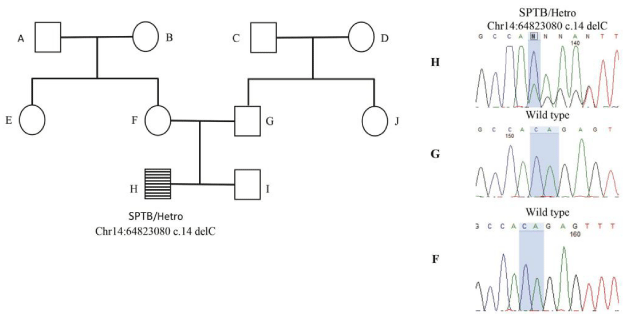
Figure 2: Genetic genealogy of family 364002.
In family 3 (family 3364803, (the proband was a 5 YO girl with early neonatal onset of severe jaundice, pallor, anemia and hemolysis who received blood transfusion at one month of age due to clinical diagnosis of HS disease. Anemia was reported in her half-sibling and paternal cousins, suspected to thalassemia minor (samples were not available to test). Recurrent urinary tract infection was reported in the affected case from 1 month. DMSA renal scan showed left poles damage with no signs of nephrolithiasis and hydronephrosis in renal ultrasound study. In addition, proband did not present with auditory deficits or cognitive impairment. Her parents were consanguineous and had no splenomegaly, renal failure or bone disease in the time study. There was some hematologic presentation of HS including peripheral blood spherocytes in PBS and increased osmotic fragility test without any frank presentation of anemia in her mother. Clinical feature of the proband and the pedigree were shown in Table S1 and Figure 3 respectively. Two different Variants of Uncertain Significance (VUS) in SPTA1 and SLC4A1 genes were shown in the proband (Table 1). Segregation analysis in the family (Sanger sequencing of the proband and her parents) showed that the homozygote variant in SLC4A1 gene (c.2494C>T) in the affected case, was inherited from heterozygote carrier parents. The heterozygote variant in SPTA1 gene (c.466C>T) was inherited from her heterozygote mother (confirmed by Sanger sequencing in the proband and her mother (Figure 3). The presence of SLC4A1 homozygote mutation in the proband beside a history of failure to thrive and urinary infection in neonatal age was in line with the diagnosis of incomplete distal renal tubular acidosis with AR inheritance that could be more severe presented due to the co-presence of the variant in the SPTA1 gene.
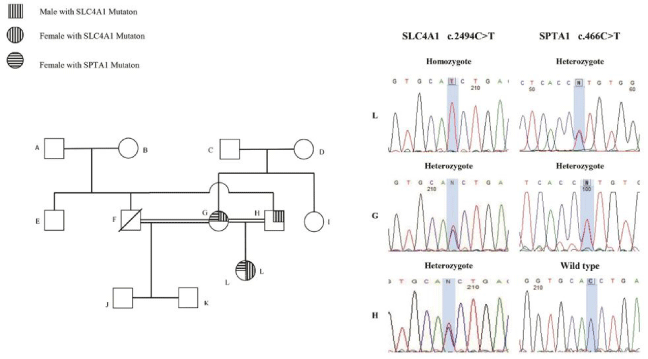
Figure 3: Genetic genealogy of family 3364803.
In family 4 (family 366364), the proband was a 8 YO boy with neonatal presentation of jaundice and pallor from 2 months old that was initially has been diagnosed as thalassemia minor (C30; AGG>ACG) that was inherited from his father with anemia, icter and reticulocytosis. Clinical feature and the pedigree were shown in Table S1 and Figure 4 respectively. As the clinical presentation couldn’t be explained with thalassemia minor, whole exome analysis was made for the affected case, his parents and uncle (Trio-plus). Two heterozygous Variants of Uncertain Significance (VUS), in KIF22 (c.1563_1564delAG) and NOTCH3 genes (c.2647C>T) have been detected in the proband that were absent in the proband’s parents (confirmed by Sanger sequencing). We also detected two different heterozygote variants of uncertain significance (VUS), in EPB41 (c.1228G>T) and PIEZO1 genes (c.2857C>T) in the proband’s father that were absent in the proband and his mother (confirmed by Sanger sequencing) (Table 1).
Figure 4: Genetic genealogy of family 366364.
In family 5 (family 318635), the proband was a 5 YO girl with microcytic hypochromic anemia who receive monthly blood transfusion from 3 months old with clinical diagnosis of thalassemia major. There was a family history of thalassemia major in her maternal cousin (IVSI-5 homozygote). Her mother was known carrier of thalassemia minor (IVSI-5 heterozygote, known mutation) and her brother also receives blood transfusion with similar manifestation. Clinical feature and the pedigree were shown in Table S1 and Figure 5 respectively. Molecular studies revealed mutation in HBB gene (IVSI- 5 heterozygote) and no other HBB gene mutation in this sibling. No deletion and duplication in HBB gene was found in the proband’s brother investigation using MLPA technique. As the primary clinical diagnosis of thalassemia major, hadn’t been confirmed, whole exome sequencing was made for the proband, her affected brother and healthy parents to investigate other genes with related to the other genetic cause of anemia like hereditary spherocytosis. We detected a previously reported homozygote missense variant (c.683G>T, NP_060345.2:p.Gly228Val) in SLC25A38 gene that was present in both affected children (homozygote) and their parents (heterozygote). This variant has been reported previously in HGMD database (CM114292) (PMID: 21393332), and likely pathogenic variant in the SLC25A38 gene was reported in sideroblastic anemia-2, pyridoxine-refractory: SIDBA2 (PMID: 21393332 and 19412 178) (OMIM # 205950, AR).
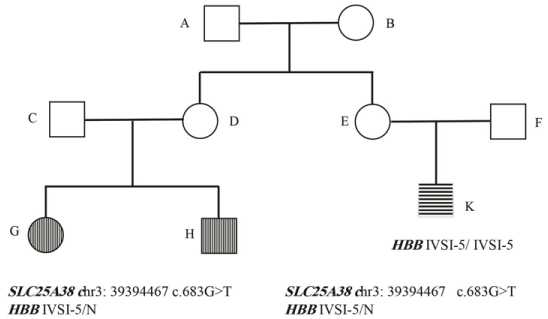
Figure 5: Genetic genealogy of family 318635.
Discussion
Hereditary Spherocytosis (HS) is a heterogeneous inherited disorder with wide range of clinical presentation and different mode of inheritance. HS originates from the mutations in the many genes coding various components of RBC membrane and molecular study of the related genes and detection of the causative variations is the mainstream of its diagnosis. Different genes are correlated to the pathogenicity of HS so far including ANK1, SPTB, SPTA1, SLC4A1, and EPB42. However some previous studies, were reported cell membrane disorders and hemolytic anemia based on clinical and laboratory characteristics in Iran [11-14], to our knowledge this the first report of molecular study of hereditary spherocytosis using Next Generation Sequencing (NGS) method in Iran. In the present study, we detected the pathogenic variation in ANK1 and SPTB in two unrelated families. In two other families, we found VUS, candidate variant that their correlation and pathogenicity hadn’t been confirmed through familial segregation study. In one case the final molecular diagnosis was sideroblastic anemia.
In family 1, the causative pathogenic variant was found in the ANK1 gene that encodes a 206-kD protein (Ankyrin protein) and is the most common cause of HS (Approximately half of all HS patients) following by SLC4A1 and SPTB genes [7]. The major function of ankyrin is to stabilize the membrane structure by interacting with beta-spectrin, protein 4.2, and band 3 protein and the deficiency of ankyrin protein leads to a decrease in spectrin assembly on the membrane skeleton thus, causes Loss of membrane [15]. Different kind of variation have been determined along the gene, including missense, nonsense, frameshift and splicing mutations (more than 60) with autosomal dominant or autosomal recessive pattern [16,17]. Ankyrin is combined of three major domains, including a multiple ankyrin repeats N-terminal domain, a spectrin-binding center region, and a regulatory C-terminal domain [18]. A de novo pathogenic variant in the SPTB gene was detected in family 2 that encodes β-spectrin protein. It plays a significant role in the strength of the erythrocyte membrane [16] and mutation in the gene may change the RBC membrane structure and surface area, and thus inducing erythrocyte sphering. This change will decrease cell deformability and lead to premature destruction in the spleen [19]. Mutations are spread all over the gene, mostly private and expected to truncated protein [20]. We detected two different Variants of Uncertain Significance (VUS) in SPTA1 (heterozygote) and SLC4A1 (homozygote) genes in the proband of the family 3 that are associated with different phenotype and mode of inheritance. SPTA1 gene is associated with eliotocytosis 2; EL2 (AD, #130600) and Spherocytosis type 3 (AR, #270970) and SLC4A1 gene with distal renal tubular acidosis (AD, #179800 and AR, #611590) and Spherocytosis type 4 (AD, #612653). These findings in the proband seem to be in line with the diagnosis of incomplete distal renal tubular acidosis (AR inheritance) that could be more severe presented due to the co-presence of the variant in the SPTA1 gene (AD inheritance). Presence of heterozygote variant in SLC4A1 in the mother cannot explain this clinical presentation as it is also present in healthy father. Mother’s mild phenotype couldn’t be explained by HE and seems to be related to HS due to the RBC osmotic fragility and spherocyte in PBS. Sequencing data analysis in family 4 detected two heterozygote Variants of Uncertain Significance (VUS) in the proband’s affected father (in EPB41 and PIEZO1 gene) that is associated with hereditary elliptocytosis (AD, #611804) and hereditary xerocytosis (AD, #194380). Segregation analysis showed that the affected father variations did not inherit to his son. Coincidental defects of two genes related to red cell membrane and/or metabolism are not very common and might be act as a modifier for the clinical presentation of the RBC membrane defect; and severity of hemolysis in cases with hereditary spherocytosis [21]. The result in this family is not conclusive and needs further phenotype genotype correlation study within the family and also further population data to classify the variants. Sequencing the proband in family 5 showed a likely pathogenic variant in SLC25A38 gene that is related to a form of anemia in which the bone marrow produces ringed sideoblasts rather than healthy peripheral erythrocytes.
In conclusion, sequencing and analysis of the multiple genes in a single test using high-throughput Next-Generation Sequencing (NGS) test, seems to act as a powerful genetic diagnostic tool and increase diagnostic efficiency [16] for congenital hemolytic anemia especially if done as parents-offspring trio test. It enables us to find the causative pathogenic variant in many cases. However, we need further familial study and population data to classify the yielded variants of uncertain significant and also genes that has not correlated to the phenotype yet. Genetic diagnosis of congenital anemia and especially hereditary spherocytosis is more complicated mainly because of the genetic heterogeneity and the large size of the genes related to the phenotype [2]. Therefore, next-generation sequencing could be a cost-effective for the molecular diagnosis of spherocytic hemolytic anemia, especially when family history is uninformative, physical examination or when routine laboratory findings cannot recognize hemolytic effect and type of inherited anemia. Molecular study of HS take advantage of assessing the risk of HS for the other relatives and also help them with their future pregnancies to have a healthy child.
Acknowledgment
Firstly, we would like to express my thanks to patients and their families for this research project. I’d also like to thank Dr. Shahla Ansari Damavandi (Ali-Asghar Children’s Hospital) for guidance on this project. We thank our colleagues at Kawsar Human Genetics Research Center (Tehran, Iran).
Highlights
• Red cell membrane disorders that affect its membrane stability results in irregular red cell shape.
• Hereditary spherocytosis is a heterogeneous inherited disorder due to red blood cell structural proteins with a wide range of clinical presentation and different mode of inheritance.
• Next-generation sequencing could be cost-effective for the molecular diagnosis of spherocytic hemolytic anemia.
References
- Tse WT, Lux SE. Red blood cell membrane disorders. British journal of haematology. 1999; 104: 2-13.
- Lin P-C, Chiou S-S, Lin C-Y, Wang S-C, Huang H-Y, Chang Y-S, et al. Whole-exome sequencing for the genetic diagnosis of congenital red blood cell membrane disorders in Taiwan. Clinica Chimica Acta. 2018; 487: 311- 317.
- Cesur M, Temiz F, Acipayam C, Kilinc M, Seringec Akkececi N. Disordered bone metabolism in hereditary spherocytosis patients. Hematology. 2019; 24: 276-281.
- Mariani M, Barcellini W, Vercellati C, Marcello AP, Fermo E, Pedotti P, et al. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. haematologica. 2008; 93: 1310-1317.
- Bolton-Maggs P, Stevens R, Dodd N, Lamont G, Tittensor P, King MJ, et al. Guidelines for the diagnosis and management of hereditary spherocytosis. British journal of haematology. 2004; 126: 455-474.
- Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood reviews. 2013; 27: 167-178.
- Park E, Phaymany V, Yi ES, Phangmanixay S, Cheong HI, Choi Y. Primary Autosomal Recessive Distal Renal Tubular Acidosis Caused by a Common Homozygous SLC4A1 Mutation in Two Lao Families. Journal of Korean medical science. 2018; 33.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. The Lancet. 2008; 372: 1411-1426.
- Agarwal AM, Nussenzveig RH, Reading NS, Patel JL, Sangle N, Salama ME, et al. Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. British journal of haematology. 2016; 174: 806-814.
- Shin S, Jang W, Kim M, Kim Y, Park SY, Park J, et al. Targeted nextgeneration sequencing identifies a novel nonsense mutation in SPTB for hereditary spherocytosis: A case report of a Korean family. Medicine. 2018; 97.
- Alavi S, Arabi N, Yazdi MK, Arzanian MT, Zohrehbandian F. Hereditary spherocytosis unmasked by human parvovirus B19 induced aplastic crisis in a family. Iranian journal of medical sciences. 2015; 40: 461-464.
- Bahrami R, Pishva N, Shahriari M, Naghshzan A. Prevalence and Assessment of the Appropriate Laboratory Indices for Screening of Hemoglobinopathyies in Southern Iranian Newborns. Iranian Journal of Neonatology IJN. 2012; 3: 63-68.
- Nazemi A, Molavi M, Raeisi E. Hereditary spherocytosis among neonates with jaundice in Bandar Abbas, South Iran. Iranian Journal of Blood and Cancer. 2015; 7: 75-78.
- Sharifzadeh S, Golafshan H, Karimi M, Ranjbaran R, Kalantari T, Moezi L. Evaluation of red cell membrane cytoskeletal disorders using a flow cytometric method in south of Iran. Laboratory & Diagnosis. 2014; 5: 23-31.
- Wang X, Mao L, Shen N, Peng J, Zhu Y, Hu Q, et al. An ANK1 IVS3-2A>C mutation causes exon 4 skipping in two patients from a Chinese family with hereditary spherocytosis. Oncotarget. 2017; 8: 113282-113286.
- Wei H, Cui Y, Lv G, Chen Y, Wang W. Analysis of Gene Mutation Related with Hereditary Spherocytosis in a Rhmod Patient of Chinese Han. J Mol Genet Med. 2018; 12: 1747.
- He B-J, Liao L, Deng Z-F, Tao Y-F, Xu Y-C, Lin F-Q. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta haematologica. 2018; 139: 60-66.
- Zou R, He X, Chen K, You Y, Zou H, Tian X, et al. A novel mutation in the ANK1 gene causes hereditary spherocytosis in a Chinese patient. Int J Clin Exp Pathol. 2017; 10: 350-358.
- Meng L-L, Yuan S-M, Tu C-F, Lin G, Lu G-X, Tan Y-Q. Next-generation sequencing identified a novel SPTB frameshift insertion causing hereditary spherocytosis in China. Ann Hematol. 2019; 98: 223-226.
- Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clinical genetics. 2016; 90: 69-78.
- Fermo E, Vercellati C, Marcello AP, Zaninoni A, van Wijk R, Mirra N, et al. Hereditary xerocytosis due to mutations in PIEZO1 gene associated with heterozygous pyruvate kinase deficiency and beta-thalassemia trait in two unrelated families. Case reports in hematology. 2017; 2017.