
Review Article
Austin J Cancer Clin Res 2014;1(2): 1008.
Current Management and Molecular Targets of Synovial Sarcoma
Lange SES1, Kremer JC1, Schenone AD1,2, Schultze MB1 and Brian A Van Tine 1,3*
1Department of Internal Medicine and Medical Oncology. Washington University, USA
2College of Medicine, Des Moines University, USA
3Siteman Cancer Center, Washington University School of Medicine, USA
*Corresponding author: Brian A Van Tine, Department of Medicine, Division of Medical Oncology, Washington University in St. Louis School of Medicine, 660 S Euclid, Campus Box 8007, St. Louis, MO 63110, USA
Received: March 17, 2014; Accepted: April 17, 2014; Published: April 21, 2014
Abstract
Synovial Sarcoma (SS) is a rare and aggressive form of soft tissue sarcoma (STS) with a high metastatic potential that is characterized by a unique translocation between SYT on chromosome 18 and SSX on chromosome X. Presently, standard of care involves surgery, radiation therapy and chemotherapy. For those patients with metastatic disease, standard of care remains enrollment in a clinical trial. While there are numerous open clinical trials for the treatment of STS in general, clinical trials designed specifically for SS remain limited. The overall low response rate to cytotoxic chemotherapies has necessitated the need for development of pathway–specific targeted therapies for SS. Deregulation of several cell signaling pathways have been identified in SS, including the SRC, Bcl–2, and MDM2 signaling pathways, which are involved with cell growth, apoptosis, and p53 regulation, respectively. Additionally, several potential enzymatic targets have been identified, including argininosuccinate synthetase 1 and histone deacetylases. Here we present an updated review of the current therapy and the prospective molecular therapeutic targets that are available for clinical trial development in SS.
Keywords: Molecular Targets; Synovial Sarcoma; Deacetylases
Introduction
Synovial Sarcoma (SS) is a rare and aggressive form of soft tissue sarcoma (STS) with a high metastatic potential that frequently develops in young people between the ages of fifteen and forty [1,2]. The incidence of SS is estimated at 900–1000 cases per year in the United States and it accounts for 8–10% of the soft tissue sarcoma patient population. Though SS is not associated with an identifiable etiologic agent or genetic predisposition, it has been associated with a gene fusion product between transcription factors SYT and SSX1, SSX2, or SSX4. This translocation has been identified in 90– 95% of all SS, and is pathognomonic and diagnostic for the disease [3]. Currently, the standard therapeutic approach to local primary disease and locally recurrent disease relies upon aggressive surgical resection, with neoadjuvant or adjuvant radiation and chemotherapy. However, in a majority of metastatic SS cases, clinical trial enrollment remains the standard of care with systemic chemotherapy remaining the sole therapeutic option off–trial. Unfortunately, the prognosis for patients presenting with metastatic disease remains poor, with a median time to cancer–specific death ranging from 10–22 months [1]. These findings highlight the need for more effective, less toxic systemic therapies for SS. In 2005, a well written work by Fukukawa et al. [4] analyzed several genes up–regulated in SS and postulated upon putative molecules for the development of novel therapies to treat SS [4]. Here, almost 10 years later, we present an updated review of the prospective molecular therapeutic targets for the treatment of SS.
Synovial Sarcoma Biology
Genetics ⁄translocation biology
SS harbors a pathognomonic chromosomal translocation t(x;18) (p11.2;q11.2) that results in a fusion between the SYT gene on chromosome 18 and one of three homologous genes (SSX1, SSX2, SSX4) on the X chromosome (Figure 1). The SYT–SSX translocation has been identified in approximately 95% of SS, and is the only cytogenetic abnormality in one–third of cases [3]. Most cases of SYT–SSX translocation associated SS harbor a fusion between SYT and SSX1 or SSX2, up to 10% of cases carry both translocations, and only rare cases carry the SYT–SSX4 translocation [5]. The anatomy of this SYT–SSX fusion oncogene has been extensively studied to better understand its pathogenicity, and it has been linked to aberrant E–cadherin repression, over expression of Bcl–2, and down–regulationof Mcl1 [6,7].
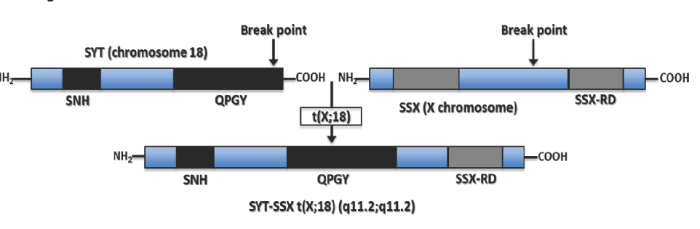
Figure 1: The SS translocation between the SYT transcriptional activator on chromosome 18 (Top Left) and the SSX transcriptional repressor located on the X chromosome (Top Right), specifically the translocation t(X;18) (p11.2;q11.2) (Bottom). The possible chromosomal rearrangements causing SS tumor genesis are SYT–SSX1, SYT–SSX2, and rarely SYT–SSX4.
The SYT gene, also reported in the literature as SS18 or SSXT, is located on chromosome 18q11 and encodes a 387–amino acidprotein. SYT is evolutionarily conserved and thought to function as a housekeeping gene given the presence of CpG islands and the absence of TATA elements in the promoter region [8]. The 54–amino acid N–terminal domain of SYT, referred to as the SNH domain, is believed to interact with SWI⁄SNF to alter chromatin remodeling and gene expression. SWI⁄SNF is a multi protein complex which counteracts repression by way of chromatin structural proteins such as histones and the polycomb–group of proteins (Figure 2) [8–11]. The C–terminal domain of SYT is rich in glutamine, proline, glycine, and tyrosine and is referred to as QPGY domain; it may function as a transcriptional activation domain on the basis of its similarity to corresponding domains in other transcriptional regulators [11].
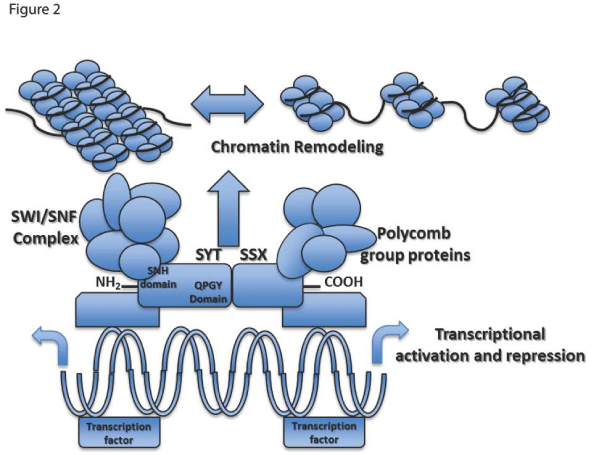
Figure 2: The SYT–SSX fusion protein alters gene expression level by modifying chromatin structure. The fusion protein associates with the SWI⁄SNF complex toaltersepigenetic regulation of a large subset of genes. Differential expression levels can be the result of deregulated histone methylation, acetylation, promoter methylation, or a number of other epigenetic mechanisms.
The structure of SSX contains two major domains found in both SSX1 and SSX2; the N–terminal Kruppel–associated box (KRAB) domain, and the C–terminal dominant repressor domain SSX–RD [11–12]. The SSX–RD domain is the most highly conserved region of the protein among the various SSX1, SSX2, and SSX4 translocations. It is also believed to be required for the nuclear co–localization with the polycomb–complex that function to repress transcription through modification of higher–order chromatin structure [13].
The chimeric transcript in SS replaces the 5’ portion of SSX with all but the eight C–terminal amino acids of SYT, but these eight amino acids do not appear to affect trans activation by the SYT QPGY domain. Together, the SYT–SSX fusion protein, in the SYT–QPGY and SSX–RD domains, displays both transcriptional activating and repressing domains, thus complicating hypotheses surrounding the oncogenicity of SYT–SSX [11]. Several studies have since demonstrated that the fusion protein is essential for tumor cell survival. Knockdown of the SYT–SSX1 protein decreases cell viability in SS; this finding of decreased viability is likely secondary to an increase in apoptosis [14–16]. Furthermore, re–introduction of a deleted exon 8 sequence into novel, patient–derived SS cell lines resulted in a decrease in cell viability as well, indicating that this splicing variant is important for the survival of SS [15].
Current Management
Although SS may develop at any anatomic site, they are found in the extremity in approximately 80% of cases, with lower extremities accounting for approximately 70% of cases [17]. In eight, seven, and five percent of cases, SS arise within the trunk, retroperitoneum⁄ abdomen, and head⁄neck, respectively [18]. Cases in the extremities may predominantly involve the very distal aspect of either the hand⁄ wrist or the foot⁄ankle and are often noted to be a palpable, slowly growing, and sometimes painful mass in the particular region of the joint. They rarely involve the actual joint, and, despite their name, are not associated with synovial tissue. Because of slow growth and insidious onset, there may be a delay in diagnosis, with one studyciting an average of 2.5 years of symptomatology before patients sought medical care [19]. Once metastatic, SS are recognized to invade both locally at the primary site and distantly to the lungs and other sites. Intriguingly, SS has been shown to have a higher risk for lymph node metastases, with an incidence of 10–12% compared to approximately 3–5% for STS in general [20].
However, although there have been many published studies investigating the natural history of the disease, it has been difficult to draw clear conclusions in regards to the prognostic factors, treatment outcomes, and survival statistics of SS in adults. This is most likely secondary to the rare incidence of the disease itself, the inclusion of children⁄adolescents in some retrospective studies, and variations in surgical and medical treatments. Although SS is viewed as moderately sensitive to chemotherapy, the 5–year distant recurrence rates, –year survival, and 10–year survival rates remain at 39%, 60% and 34%, respectively [1]. In 2000, Lewis et al published a retrospective analysis of 112 patients with localized primary extremity SS treated at Memorial Sloan–Kettering Cancer Center. These patients underwent surgical excision with curative intent, with only 22% requiring amputation. Chemotherapy and radiation therapy was administered only to 37% and 46% of patients, respectively, although treatmentwas not standardized. The 5–year recurrence rate was noted to be 39% in this cohort, with a majority of recurrences occurring in the lungs. In total, at 5 years, tumor related mortality was 25% for patients with primary extremity SS [21]. In regards to timing of recurrence, Singer et al in 1996 recognized a high rate of late metastasis, noting a 5–year survival rate of 60% and a 10–year survival rate of just 34%,with almost half of the disease–specific deaths occurring between 5 and 10 years [2]. Also, in those studies that have included patients with metastatic disease at the time of presentation, the median time to cancer–specific death ranges from 10–22 months [1,22].
Prognostic factors of disease–specific survival identified in multiple studies include age, size of tumor, margin status at resection, mitotic activity, bone or neurovascular invasion, histological subtype, p53 expression, Ki67 proliferative index, and SYT–SSX fusion type. Lesion size at presentation is variable, although most patients present with tumors larger than five centimeters. Those tumors that present at the periphery of a limb, for example, may be diagnosed earlier at a smaller size, although the rarity of the tumor and physical similarity to benign lesions may still result in a delay in diagnosis. In particular, large primary tumor size has consistently been associated with development of distant metastasis and decreased disease–specific survival [18]. Computed topography (CT) typically demonstrates a non infiltrative, well–circumscribed mass often with punctuate peripheral calcifications, although MRI is the modality of choice for diagnosis and initial staging of SS [23,24].
Pathological
Immunohistochemical and structural characteristics of SS cells are clearly different than those of synovial cells. SS have primarily beenconsidered a high–grade tumor by definition, although some groupsin Europe and Asia may classify SS as either high–grade or low–grade[25,26]. Morphologically, SS tumors can be composed of two distinct cell types in varying proportions. Each tumor typically consistsof spindle cells and⁄or epithelioid cells, allowing for classificationinto three histologic subtypes: monophasic, biphasic, and poorly differentiated (Figure 3) [27,28]. The spindle cell component of a tumor consists of small, uniform, and ovoid cells with sparse cytoplasm and understated pale nuclei and nucleoli. The monophasic histologic subtype of SS tumors display primarily spindle cells with rare if any epitheiloid cells, arranged in intersecting fascicles with a hemangiopericytoma–like vascular pattern that often includes calcifications. Two–thirds of cases are of monophasic subtype [19].
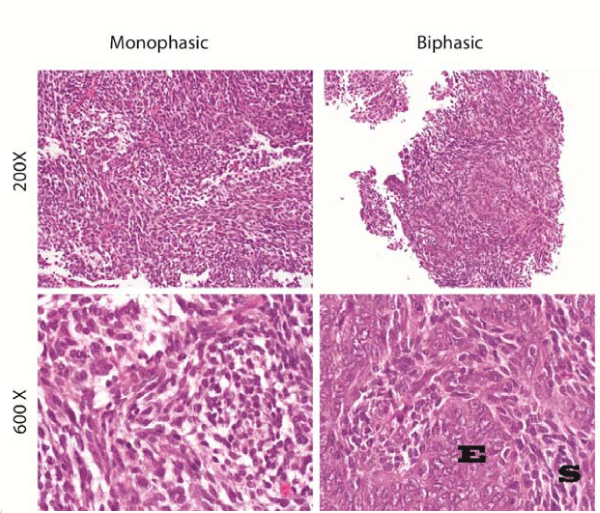
Figure 3: Left column demonstrates monophasic synovial sarcoma at 200X (Top) and 600X (Bottom). Right column demonstrates biphasic synovial sarcoma at 200X (Top) and 600X (Bottom). E marks the epithelial component and S marks the spindle component of the biphasic SS.
In contrast, biphasic tumors consist of both spindle and epitheliod cell types, with the epithelioid component displaying cells with abundant cytoplasm and ovoid nuclei that often form glandular structures. The epithelioid component, when in glandular formation, displays features typical of adenocarcinoma; for example, luminacontaining epithelial mucin or papillary structures [27]. When monophasic tumors display the epithelioid subtype, cytogenetic analysis is usually required for diagnosis, as these tumors can be otherwise indistinguishable from adenocarcinoma [29]. Finally, the poorly differentiated histologic subtype of SS can be difficult to distinguish from other high–grade small–cell tumors, as they display rhabdoid features, dense cellularity, numerous mitotic figures, and areas of necrosis that canbe extensive [30].
Often, SS can be difficult to diagnose with routine histologic examination. Approximately 90% of tumors are cytokeratin–positive, with the epithelioid cell type staining stronger than the spindle cell type. Although SS are histologically similar to other spindle cellsarcomas, they stain positive for cytokeratin 7 and 19, which allows for reliable distinction between SS and primitive neuroectodermal and malignant peripheral nerve sheath tumors [31, 32]. Epithelial membrane antigens, which are expressed in SS but not in other spindle cell sarcomas, and Bcl–2, which is upregulated in SS, may also assist in immunohistochemical staining for diagnostic purposes [32]. Additionally, SS tumors stain positive for vimentin, supporting the hypothesis that SS cells are derived from multipotent stem cells of a mesenchymal and⁄or epithelial origin, rather than from synovial tissue as their moniker would suggest [33].
Molecular⁄Cytogenetic diagnosis
As SS can be difficult to diagnose through current clinical and histopathologic analysis, molecular testing should also be performed in cases with low to moderate clinical suspicion of SS [18]. Most biphasic tumors contain SS18–SSX1 while almost all tumors with SS18–SSX2 are monophasic. However, monophasic tumors have an equal chance of containing either SS18–SSX1 or SS18–SSX2 [34]. A tumor with a SS18–SSX translocation may be diagnosed via fluorescence in situ hybridization (FISH) or reverse transcriptasepolymerase chain reaction (RT–PCR) [29]. These diagnostic modalities may be performed on frozen or paraffin–embedded clinical samples [35,36,37].
Current Therapy
Surgery
The foundation for treatment of STS, as well as SS, is complete surgical resection with wide margins [18]. Standard of treatment dictates that non–curative surgery, such as an intra–lesion resection or incomplete gross resection should not be offered or attempted. As SS are most commonly diagnosed in an extremity and in proximity to a joint, careful dissection of tissue planes and neurovascular structures is necessary. Fortunately, it is uncommon that amputation of an extremity is required as primary surgical therapy [19]. Of those patients who undergo surgery for locally recurrent disease, limbsparing surgery is possible for the majority of patients, although greater morbidity involving critical structures like nerves, veins, arteries, or even bone may be expected [19]. Even in cases involving a limb, amputation is still considered a last resort [38].
Surgical therapy for patients with metastatic disease requires careful patient selection to ensure benefit; considerations include patient performance status, extent of disease burden, disease–free interval, and response to systemic chemotherapy. For those patients with pulmonary metastasis, complete resection has been shown to improve survival [39].
Radiation therapy
Radiation therapy has been employed for SS in the neoadjuvant and the adjuvant setting. Preoperative radiation therapy in the form of external beam radiation is used to reduce tumor size at resection, but is associated with reduced dose, smaller fields, and a higher risk of long–term wound complications [19]. Radiation is typically usedin the adjuvant setting for patients with tumors over five centimetersor for areas where local tumor control may be difficult, as in the headand neck. At this time, the use of concurrent systemic chemotherapy and radiation is investigational [19].
In patients who experience local recurrence, the re–irradiation of a site is typically not feasible, as those patients have received maximum radiation dosage to that site. However, the use of brachytherapy maybe possible with minimal morbidity in patients with prior radiation. A group at Memorial Sloan–Kettering Cancer Center has reported the largest experience with the use of brachy therapy and interstitial afterloading brachytherapy. In a study investigating the local control of surgery and brachytherapy compared to surgery alone, local controlwas 95% versus 54%, significantly higher in those who received brachy therapy [40]. In a study from 1990, Nori reported an overall 5–year actuarial local control of 68% with the use of interstitial brachytherapy; however, this study included patients with various types of recurrent sarcomas who experienced varying numbers of recurrences [41].
Chemotherapy
SS are considered to be moderately chemotherapy sensitive, and are among the more chemotherapy sensitive STS. Anthracyclinebased chemotherapy employing doxorubicin was the first chemotherapy agent to display activity against SS, although response rates of doxorubicin combinations approach 45% at 10 years, there is no statistically significant difference in overall survival noted compared to sequential therapy [42].
For those patients who present with metastatic disease, clinical trial enrollment is considered the standard of care. For the treatment of metastatic disease off trial, ifosfamide, anthracyclines and pazopanib are the most active agents. Some physicians choose to increase the dose of ifosfamide in the setting of recurrence if the patient has been previously treated with an ifosfamide regimen [19].
Pazopanib is an oral angiogenesis inhibitor that targets vascular endothelial growth factor receptors (VEGFRs), platelet–derived growth factor receptors (PDGFRs), and c–kit. In a phase II study from the European Organization for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group (EORTC Study 62043) in which 37 SS patients were enrolled [43], progression free rate at twelve weeks of treatment was reported as the primary end point, and 49% of patients with SS demonstrated stable disease. In 2012, a multi–center phase III study also designed by EORTC, 369 patients with angiogenesis inhibitor–naive, metastatic STS, progressing despite previous standard chemotherapy, were randomly assigned to receive pazopanib or placebo. Median progression–free survival for SS patients was longer for pazopanib compared with placebo (4.1 months versys 0.9 months) [44].
Current Clinical Trials
As no targeted therapy for SS has yet to be developed, enrollment in clinical trials is recommended as standard of care at this time. For patients newly diagnosed with all subtypes of STS, there are many clinical trials enrolling at this time. The number of open clinical trials specific to SS, however, remains limited. A phase I study of genetically engineered NY–ESO–1 specific (c259) T cells in HLA–A2+ patients with SS opened, with the purpose of testing the effects of chemotherapy and the NYESO T cells on patients with metastatic and recurrent SS [45].
The potential of NY–ESO–1 as a cancer therapeutic target relies on the concept of specific immune recognition of cancer, and the subsequent development of an anti–cancer response. The NY–ESO–1 gene is found on the X chromosome at q28, and it codes for several products, namely NY–ESO–1, a 180–amino acid protein. Expression pattern analysis by RT–PCR for NY–ESO–1 has confirmed that the protein in normal tissue is restricted to testis, but found frequently in cancer. The function of the protein is yet to be determined; however, the presence of NY–ESO–1 is noted in one–third to one–fourth of all melanoma, lung, esophageal, liver, gastric, prostate, ovarian, or bladder cancers [46]. Strikingly, approximately 80% of SS have been found to express NY–ESO–1 [47], which holds promise for immunotherapeutic approaches such as NY–ESO–1 specific T cells as noted above. In this phase I study, the primary outcome measure is to determine whether the administration of T cells genetically engineered to recognize a peptide derived from NY–ESO–1 in HLA–A2+patients demonstrate a response rate consistent with that observed using similar NYESO–1 specific T cells plus aldesleukin in patients with SS. This trial is open to patients who are HLA–A2+ with SS that have been treated with standard chemotherapy with remaining measurable disease that is metastatic, progressive⁄ persistent, recurrent, or unresectable. In September of 2013, the NCI opened a second clinical trial for enrollment involving NY–ESO–1 in order to determine whether the administration of anti–ESO mTCRengineered peripheral blood lymphocytes plus high–dose aldesleukin following a non–myeloablative lymphoid depleting regimen will result in objective tumor regression in patients with metastatic cancers that express the ESO antigen, including SS [45].
Onco Therapy Science Inc, opened a clinical trial in France for patients with histologically confirmed progressive SS that is resistant to doxorubicin and ifosfamide in order to investigate a chimeric humanized monoclonal antibody against FZD10, named OTSA101. The gene encoding frizzled homologue 10 (FZD10), a 7–transmenbrane receptor and member of the Wnt signalling receptor family, is overexpressed in SS and is undetectable in normal humanc tissues except placenta. Non–radio labeled OTSA101 antibody has only weak antagonistic activity on SS cell growth in vivo. However, Yttrium 90–radiolabeled OTSA101 (OTSA101–DTPA–90Y) showed significant antitumor activity following a single intravenous injection in mouse xenograft model. This study should allow for definition of the optimal recommended dose of this novel monoclonal antibody therapy [48].
Molecular Targets for Clinical Trials
SRC: c–SRC (SRC) is a non–receptor tyrosine kinase involved in regulation of cell growth, survival, and motility [49]. The kinase has two phosphorylation sites, with phosphorylation of Tyr527 reducing kinase activity, and autophosphorylation of Tyr416 inducing full kinase activity [50,51]. SRC phosphorylation at Tyr527 is regulated by c–SRC tyrosine kinase (CSK) and the protein tyrosine phosphatase PTP1B [52,53]. Deregulation of CSK, as well as overexpression of PTP1B, have been shown to be critical in a number of cancers, including colon cancer and breast cancer [54–56]. SRC has also been shown to interact with a number of receptor tyrosine kinases, including insulin like growth factor–1 receptor (IGF–1R), and effectors of the PI3K⁄AKT, RAS⁄MAPK, and STAT3 pathways. SRC interaction with focal adhesion kinase (FAK), various integrins, and regulators of the Rho–GTPases allows regulation of cellular migration [57–61].
Deregulation of SRC signaling is crucial for growth and survival of SS cells. SRC Tyr416 phosphorylation has been shown to be one of the strongest phosphorylated kinases in SS cell lines [49]. The high level of Tyr416 phosphorylation is induced by the SS translocation SYT–SSX, which has been shown to up–regulate IGF–1R, EGFR, and Her2 signaling [58,62–64].
The SRC inhibitor dasatinib has been shown to inhibit growth in SS cell lines, as well as increase apoptosis and decrease the mitotic rate in SS cells. Combination of dasatinib with conventional chemotherapy drugs has shown additive effects in SS cell lines [49]. Inhibitors of IGF–1R, EGFR, or Her2 signaling may also be suitable targeted therapies for SS due to their ability to decrease SRC signaling (Figure 4).
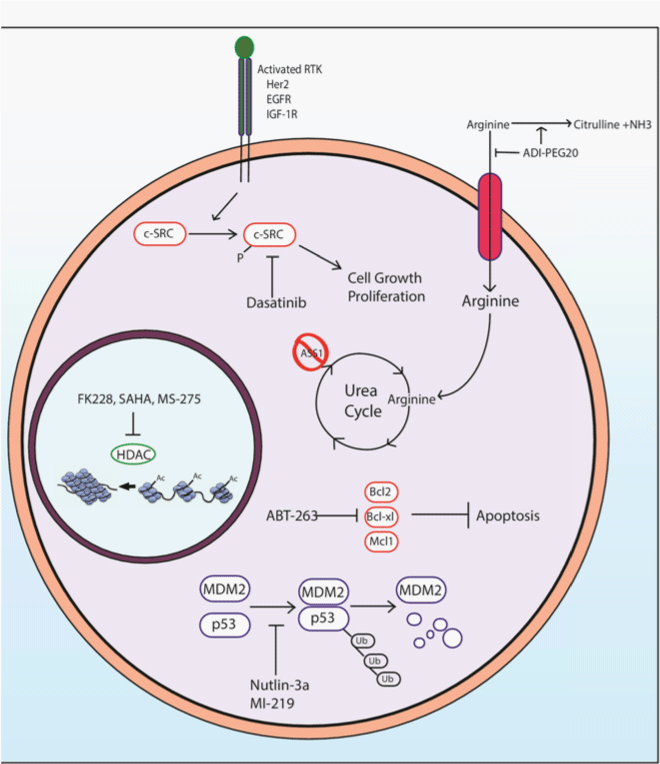
Figure 4: A schematic of the possible therapeutic targets for SS. Reliance upon SRC signaling suggests inhibition of upstream receptor tyrosine kinases as well as SRC itself can provide therapeutic benefits. The epigenetic silencing of ASS1 can be targeted via depletion of extracellular arginine. As SS utilizes Bcl–2 over expression in order to increase resistance to apoptosis, targeting with Bcl–2 inhibitors will induce apoptosis in tumor cells. MDM2 amplification causes p53 polyubiquitination and proteasomal degradation, thus inhibition of the MDM2–p53 interaction can increase downstream p53 signaling. Lastly, inhibition of HDACs has been shown to increase expression of tumor suppressor genes silenced via histone deacetylation.
Bcl–2: The Bcl–2 protein family is important in regulation of apoptosis, and frequently found to be mutated in a number of cancers. Bcl–2 is one of the antiapoptotic members of the family, along with Mcl–1, Bcl–xl, and a number of other proteins [65,66]. The family includes apoptosis promoting proteins, including Bad, Bax, and Bid, among others. By alternatively regulating the permeability of the mitochondrial membrane, the proapoptotic Bcl–2 proteins can cause cytochrome c release from the mitochondria and subsequent initiation of apoptosis. The BH3 domain is the ‘death domain’, found in all proapoptotic Bcl–2 proteins and capable of introducing a pore into the mitochondrial membrane to allow cytochrome c release.
SS has deregulated expression of a number of proteins of the Bcl–2 family, including both antiapoptotic and proapoptotic proteins [6]. Part of the SS expression signature includes high level of Bcl–2 expression, leading to apoptotic resistance [67–69]. Bclxl, another antiapoptotic protein, has also been shown to be up regulated by the SYT–SSX fusion protein. Mcl–1 and Bcl2a1a, both antiapoptotic proteins, are down regulated by SYT–SSX expression [6]. Up regulation of anti–apoptotic proteins likely contributes to the resistance of SS to many cytotoxic chemotherapy.
Targeted therapeutics directed towards the apoptotic pathway have been developed capitalizing on the BH3 death domain of the Bcl–2 proapoptotic proteins. BH3 domain peptidomimetics have been shown to be capable of targeting SS cell lines and xenografts and inducing apoptosis. ABT–263 is a potent Bcl–2 inhibitor and capable of sensitizing SS to classical cytotoxic chemotherapies [6] (Figure 4). The lack of Mcl–1 expression, the natural therapeutic bypass to the Bcl–2 inhibitors, makes Bcl–2 a highly attractive target for the treatment of SS.
MDM2: MDM2, or mouse double minute 2 homolog, is a key protein in the p53 signaling pathway. MDM2 functions as an E3 ubiquitin ligase [70]. Under normal cellular conditions, MDM2 binds to and poly–ubiquitinates p53, marking the transcription factor for proteasomal degradation. When the cell experiences a p53 activating signal, including DNA damage, oxidative stress, or ribonucleotide depletion, MDM2 function can be inhibited by a number of mechanisms to up regulate p53 function and subsequently up regulate p53 target genes [71]. MDM2 can be inhibited by phosphorylation or localization to the nucleolus by p14arf, the protein product of the alternative reading frame of the p16 INK4a locus. Activation of p53 can cause transcription of genes causing cell cycle arrest or apoptosis depending on the strength and duration of the activating signal [72].
While SS are typically wild type for p53, an up regulation of MDM2 has been documented [73]. The SYT–SSX fusion protein is capable of increasing p53 poly–ubiquitination and subsequent proteasomal degradation by increasing the stability of MDM2. MDM2 autoubiquitination is inhibited in SS, leading to increased MDM2 stability [74]. The increase of MDM2, and subsequent decrease of p53, leads to an increase in the ability of SS cells to survive typical apoptotic signals by attenuating the response of the p53 activating signal. The decreased level of p53 leads to the increased resistance of apoptosis in response to genotoxic stress in SS.
Since SS is typically wild type for p53, therapeutics targeting the activity of MDM2 can be particularly useful in treatment of SS due to their ability to reactivate the p53 signaling axis [75]. Small molecule inhibitors of MDM2 in combination with cytotoxic chemotherapy may provide enhanced toxicity to SS [72,76]. Two such small molecule inhibitors of this interaction include nutlin–3a and MI–219, both of which have been shown to decrease MDM2–p53 interaction, increasing the transcriptional activation potential of p53 [70] (Figure 4).
ASS1: ASS1, or argininosuccinate synthetase 1, is a key enzyme in the urea cycle, catalyzing the formation of argininosuccinate from citrulline and aspartate. Cells that are deficient in ASS1 expression rely upon arginine from the environment. When deprived of arginine, these cells are incapable of producing the amino acid and undergo autophagy [77,78]. It has been shown that in nearly 90% of sarcomas, the ASS1 promoter is heavily methylated and expression is subsequently silenced [79]. ASS1 methylation has been shown to correlate with reduced metastasis free survival. These tumors have been shown to be arginine auxotrophs, with inhibited growth and induction of autophagy upon arginine deprivation [80,81].
SS have been shown to be one of the classes of sarcoma in which ASS1 is silenced [9]. The reliance upon extracellular arginine can be exploited with use of ADI–PEG20. This pegylated form of arginine deiminase is capable of converting extracellular arginine into citrulline and ammonia and has been shown to decrease arginine levels in the blood stream and cause ASS1 deficient tumors to undergo autophagy. ADI–PEG20 has also been shown to increase the radio sensitivity of ASS1 deficient tumors, and thus may increase the susceptibility of SS to traditional radio therapy [82] (Figure 4).
HDAC: Histone deacetylases, or HDACs, are important chromatin remodeling enzymes involved in the epigenetic regulation of the genome. Histone tail acetylation decreases the electrostatic interaction between the histone tail and the DNA backbone, allowing the two to dissociate, enabling transcription. Thus, acetylation of histones correlates with increased gene expression, while deacetylation correlates to tighter associations between DNA and histones, condensation of the chromatin, and subsequently decreased gene expression. Epigenetic down regulation of tumor suppressors have been shown to contribute to SS cell growth and proliferation.
As SYT–SSX is a chromatin remodeling fusion protein, targeting this epigenetic deregulation has been shown to inhibit SS cell growth in vitro and in vivo [83]. The HDAC inhibitors have also been shown to induce apoptosis in SS cell lines. HDAC inhibitors are capable of driving the re–expression of genes typically silenced in tumors, including the p21⁄WAF1 cyclin dependent kinase inhibitor [84–86]. The HDAC inhibitors that are currently in clinical trials for cancer treatment, including FK228, MS–275, SAHA, and PXD101 could prove to be very effective in the targeted treatment of SS [87–89] (Figure 4).
Summary
The standard of care for the treatment of sarcoma is clinical trial when surgical resection is not possible. The unique biology of SS driven by its hybrid transcription factor SYT–SSX makes it ideal for histology driven clinical trials. Optimal candidates for therapy include the HDACs, SRC, MDM2, ASS1, and Bcl–2 (Figure 4, Table 4). The most likely successful strategy will involve targeting more than one of these unique SS inhibited targets simultaneously. Combinations such as MDM2 inhibition with an HDAC inhibitor or a Bcl–2 inhibitor with ADI–PEG20 and the various combinations that are possible wait direct testing in SS cell lines and patients. Rationally designed therapies based on the specific biology of SS should lead to a bright future for the treatment of SS.
Acknowledgement
The author would like to thank Ian Hagemann, M.D., Ph.D. for the use of his micrographs of SS.
References
- Ferrari A, Gronchi A, Casanova M, Meazza C, Gandola L, Collini P, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004; 10: 627-634.
- Singer S, Baldini EH, Demetri GD, Fletcher JA, Corson JM. Synovial sarcoma: prognostic significance of tumor size, margin of resection, and mitotic activity for survival. J Clin Oncol. 1996; 14: 1201-1208.
- Sandberg A, Bridge J. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: alveolar soft part sarcoma. Cancer Genet Cytogenet. 2002; 136: 1-9.
- Fukukawa C, Nakamura Y, Katagiri T. Molecular target therapy for synovial sarcoma. Future Oncol. 2005; 1: 805-812.
- Yang K, Lui WO, Xie Y, Zhang A, Skytting B, Mandahl N, et al. Co-existence of SYT-SSX1 and SYT-SSX2 fusions in synovial sarcomas. Oncogene. 2002; 21: 4181-4190.
- Jones KB, Su L, Jin H, Lenz C, Randall RL, Underhill TM, et al. SS18-SSX2 and the mitochondrial apoptosis pathway in mouse and human synovial sarcomas. Oncogene. 2013; 32: 2365-2371.
- Saito T, Nagai M, Ladanyi M. SYT-SSX1 and SYT-SSX2 interfere with repression of E-cadherin by snail and slug: a potential mechanism for aberrant mesenchymal to epithelial transition in human synovial sarcoma. Cancer Res. 2006; 66: 6919-6927.
- Haldar M, Randall RL, Capecchi MR. Synovial sarcoma: from genetics to genetic-based animal modeling. Clin Orthop Relat Res. 2008; 466: 2156-2167.
- Frith AE, Hirbe AC, Van Tine BA. Novel pathways and molecular targets for the treatment of sarcoma. Curr Oncol Rep. 2013; 15: 378-385.
- Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013; 153: 71-85.
- Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001; 20: 5755-5762.
- Lim FL, Soulez M, Koczan D, Thiesen HJ, Knight JC. A KRAB-related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas. Oncogene. 1998; 17: 2013-2018.
- dos Santos NR, de Bruijn DR, van Kessel AG. Molecular mechanisms underlying human synovial sarcoma development. Genes Chromosomes Cancer. 2001; 30: 1-14.
- Takenaka S, Naka N, Araki N, Hashimoto N, Ueda T, Yoshioka K, et al. Downregulation of SS18-SSX1 expression in synovial sarcoma by small interfering RNA enhances the focal adhesion pathway and inhibits anchorage-independent growth in vitro and tumor growth in vivo. Int J Oncol. 2010; 36: 823-831.
- Carmody Soni EE, Schlottman S, Erkizan HV, Uren A, Toretsky JA. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin Orthop Relat Res. 2014; 472: 874-882.
- Peng C, Guo W, Yang Y, Zhao H. Downregulation of SS18-SSX1 expression by small interfering RNA inhibits growth and induces apoptosis in human synovial sarcoma cell line HS-SY-II in vitro. Eur J Cancer Prev. 2008; 17: 392-398.
- Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995; 164: 395-402.
- Eilber FC, Dry SM. Diagnosis and management of synovial sarcoma. J Surg Oncol. 2008; 97: 314-320.
- Brennan, Murray F, Antonescu, Cristina R, Maki, Robert G. Management of soft tissue sarcoma. 2013; 15: 380.
- Skinner KA, Eilber FR. Soft tissue sarcoma nodal metastases: biologic significance and therapeutic considerations. Surg Oncol Clin N Am. 1996; 5: 121-127.
- Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, Woodruff JM, et al. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000; 18: 2087-2094.
- Spurrell EL, Fisher C, Thomas JM, Judson IR. Prognostic factors in advanced synovial sarcoma: an analysis of 104 patients treated at the Royal Marsden Hospital.Ann Oncol. 2005; 16: 437-444.
- Nakanishi H, Araki N, Sawai Y, Kudawara I, Mano M, Ishiguro S, et al. Cystic synovial sarcomas: imaging features with clinical and histopathologic correlation. Skeletal Radiol. 2003; 32: 701-707.
- Bakri A, Shinagare AB, Krajewski KM, Howard SA, Jagannathan JP, Hornick JL, et al. Synovial sarcoma: imaging features of common and uncommon primary sites, metastatic patterns, and treatment response. AJR Am J Roentgenol. 2012; 199: W208-215.
- Guillou L, Benhattar J, Bonichon F, Gallagher G, Terrier P, Stauffer E, et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol. 2004; 22: 4040-4050.
- Hasegawa T, Yamamoto S, Yokoyama R, Umeda T, Matsuno Y, Hirohashi S. Prognostic significance of grading and staging systems using MIB-1 score in adult patients with soft tissue sarcoma of the extremities and trunk. Cancer. 2002; 95: 843-851.
- Sharon W. Weiss, Hector Battifora, Richard L Kempson, William R Hart. Tumors of soft tissue: based on the proceedings of the 52nd Annual Anatomic Pathology Slide Seminar of the American Society of Clinical Pathologists, October 2 and 3, 1986, Orlando Marriott Hotel, Orlando, Florida. Chicago: American Society of Clinical Pathologists Press. 1987.
- Goldblum, John R, Folpe, Andrew L Weiss, Sharon W, Enzinger, Franz M. Enzinger and Weiss's soft tissue tumors. 6th edn. Philadelphia PA: Saunders/Elsevier. p. 2014.
- Coindre JM, Pelmus M, Hostein I, Lussan C, Bui BN, Guillou L. Should molecular testing be required for diagnosing synovial sarcoma? A prospective study of 204 cases. Cancer. 2003; 98: 2700-2707.
- He R, Patel RM, Alkan S, Hammadeh R, Weiss SW, Goldblum JR, et al. Immunostaining for SYT protein discriminates synovial sarcoma from other soft tissue tumors: analysis of 146 cases. Mod Pathol. 2007; 20: 522-528.
- Folpe AL, Schmidt RA, Chapman D, Gown AM. Poorly differentiated synovial sarcoma: immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol. 1998; 22: 673-682.
- Pelmus M, Guillou L, Hostein I, Sierankowski G, Lussan C, Coindre JM. Monophasic fibrous and poorly differentiated synovial sarcoma: immunohistochemical reassessment of 60 t(X;18)(SYT-SSX)-positive cases. Am J Surg Pathol. 2002; 26: 1434-1440.
- Smith ME, Fisher C, Wilkinson LS, Edwards JC. Synovial sarcoma lack synovial differentiation. Histopathology. 1995; 26: 279-281.
- Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998; 338: 153-160.
- Tsuji S, Hisaoka M, Morimitsu Y, Hashimoto H, Shimajiri S, Komiya S. Detection of SYT-SSX fusion transcripts in synovial sarcoma by reverse transcription-polymerase chain reaction using archival paraffin-embedded tissues. Am J Pathol. 1998; 153: 1807-1812.
- Shipley J, Crew J, Birdsall S, Gill S, Clark J, Fisher C, et al. Interphase fluorescence in situ hybridization and reverse transcription polymerase chain reaction as a diagnostic aid for synovial sarcoma. Am J Pathol. 1996; 148: 559-567.
- Nagao K, Ito H, Yoshida H. Chromosomal translocation t(X;18) in human synovial sarcomas analyzed by fluorescence in situ hybridization using paraffin-embedded tissue. Am J Pathol. 1996; 148: 601-609.
- Eilber FC, Brennan MF, Riedel E, Alektiar KM, Antonescu CR, Singer S. Prognostic factors for survival in patients with locally recurrent extremity soft tissue sarcomas. Ann Surg Oncol. 2005; 12: 228-236.
- Gadd MA, Casper ES, Woodruff JM, McCormack PM, Brennan MF. Development and treatment of pulmonary metastases in adult patients with extremity soft tissue sarcoma. Ann Surg. 1993; 218: 705-712.
- Brennan MF. The management of soft tissue sarcomas. Br J Surg. 1984; 71: 964-967.
- Nori D, Schupak K, Shiu MH, Brennan MF. Role of brachytherapy in recurrent extremity sarcoma in patients treated with prior surgery and irradiation. Int J Radiat Oncol Biol Phys. 1991; 20: 1229-1233.
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet. 1997; 350: 1647-1654.
- Sleijfer S, Ray-Coquard I, Papai Z, Le Cesne A, Scurr M, Schöffski P, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol. 2009; 27: 3126-3132.
- van der Graaf WT1, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012; 379: 1879-1886.
- Mackall CGS. A Pilot Study of Genetically Engineered NY-ESO-1 Specific (c259) T Cells in HLA-A2+ Patients With Synovial Sarcoma. 2013.
- Gnjatic S, Nishikawa H, Jungbluth AA, Güre AO, Ritter G, Jäger E, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006; 95: 1-30.
- Jungbluth AA, Chen YT, Stockert E, Busam KJ, Kolb D, Iversen K, et al. Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues. Int J Cancer. 2001; 92: 856-860.
- Blay JY. First in Man Study Investigating the Biodistribution, the Safety and Optimal Recommended Dose of a New Radiolabelled Monoclonal Antibody Targeting Frizzled Homolog 10 (FZD10) in Patients With Relapsed or Refractory Non Resectable Synovial Sarcomas. 2014.
- Michels S, Trautmann M, Sievers E, Kindler D, Huss S, Renner M, et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res. 2013; 73: 2518-2528.
- Roskoski R Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005; 331: 1-14.
- Bjorge JD, Jakymiw A, Fujita DJ. Selected glimpses into the activation and function of Src kinase. Oncogene. 2000; 19: 5620-5635.
- Cole PA, Shen K, Qiao Y, Wang D. Protein tyrosine kinases Src and Csk: a tail's tale. Curr Opin Chem Biol. 2003; 7: 580-585.
- Sirvent A, Bénistant C, Pannequin J, Veracini L, Simon V, Bourgaux JF, et al. Src family tyrosine kinases-driven colon cancer cell invasion is induced by Csk membrane delocalization. Oncogene. 2010; 29: 1303-1315.
- Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000; 275: 41439-41446.
- Kunte DP, Wali RK, Koetsier JL, Hart J, Kostjukova MN, Kilimnik AY, et al. Down-regulation of the tumor suppressor gene C-terminal Src kinase: an early event during premalignant colonic epithelial hyperproliferation. FEBS Lett. 2005; 579: 3497-3502.
- Zhu S, Bjorge JD, Fujita DJ. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 2007; 67: 10129-10137.
- Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res. 2006; 98: 394-402.
- Xie Y, Skytting B, Nilsson G, Brodin B, Larsson O. Expression of insulin-like growth factor-1 receptor in synovial sarcoma: association with an aggressive phenotype. Cancer Res. 1999; 59: 3588-3591.
- Friedrichs N, Trautmann M, Endl E, Sievers E, Kindler D, Wurst P, et al. Phosphatidylinositol-3'-kinase/AKT signaling is essential in synovial sarcoma. Int J Cancer. 2011; 129: 1564-1575.
- Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007; 67: 2800-2808.
- Huveneers S, Danen EH. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009; 122: 1059-1069.
- Ptaszynski K, Szumera-Cieckiewicz A, Zakrzewska K, Tuziak T, Mrozkowiak A, Rutkowski P. Her2, EGFR and TOPIIA gene amplification and protein expression in synovial sarcoma before and after combined treatment. Pol J Pathol. 2009; 60: 10-18.
- Friedrichs N, Küchler J, Endl E, Koch A, Czerwitzki J, Wurst P, et al. Insulin-like growth factor-1 receptor acts as a growth regulator in synovial sarcoma. J Pathol. 2008; 216: 428-439.
- Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000; 19: 5606-5613.
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9: 47-59.
- Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008; 27: 6398-63406.
- Hirakawa N, Naka T, Yamamoto I, Fukuda T, Tsuneyoshi M. Overexpression of bcl-2 protein in synovial sarcoma: a comparative study of other soft tissue spindle cell sarcomas and an additional analysis by fluorescence in situ hybridization. Hum Pathol. 1996; 27: 1060-1065.
- Sun B, Sun Y, Wang J, Zhao X, Wang X, Hao X. Extent, relationship and prognostic significance of apoptosis and cell proliferation in synovial sarcoma. Eur J Cancer Prev. 2006; 15: 258-265.
- Knösel T, Heretsch S, Altendorf-Hofmann A, Richter P, Katenkamp K, Katenkamp D, et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases. Eur J Cancer. 2010; 46: 1170-1176.
- Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013; 13: 83-96.
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002; 2: 594-604.
- Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009; 49: 223-241.
- Oda Y, Sakamoto A, Satio T, Kawauchi S, Iwamoto Y, Tsuneyoshi M. Molecular abnormalities of p53, MDM2, and H-ras in synovial sarcoma. Mod Pathol. 2000; 13: 994-1004.
- D'Arcy P, Maruwge W, Ryan BA, Brodin B. The oncoprotein SS18-SSX1 promotes p53 ubiquitination and degradation by enhancing HDM2 stability. Mol Cancer Res. 2008; 6: 127-138.
- D'Arcy P, Ryan BA, Brodin B. Reactivation of p53 function in synovial sarcoma cells by inhibition of p53-HDM2 interaction. Cancer Lett. 2009; 275: 285-292.
- Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor Nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Des. 2008; 14: 2100-2110.
- Lan J, Tai HC, Lee SW, Chen TJ, Huang HY, Li CF. Deficiency in expression and epigenetic DNA Methylation of ASS1 gene in nasopharyngeal carcinoma: negative prognostic impact and therapeutic relevance. Tumour Biol. 2014; 35: 161-169.
- Delage B, Luong P, Maharaj L, O'Riain C, Syed N, Crook T, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis. 2012; 3: e342.
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010; 126: 2762-2772.
- Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des. 2008; 14: 1049-1057.
- Savaraj N, You M, Wu C, Wangpaichitr M, Kuo MT, Feun LG. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med. 2010; 10: 405-412.
- Park H, Lee JB, Shim YJ, Shin YJ, Jeong SY, Oh J, et al. Arginine deiminase enhances MCF-7 cell radiosensitivity by inducing changes in the expression of cell cycle-related proteins. Mol Cells. 2008; 25: 305-311
- Su L, Cheng H, Sampaio AV, Nielsen TO, Underhill TM. EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene. 2010; 29: 4352-4361.
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006; 5: 769-784.
- Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997; 57: 3697-3707.
- Archer SY, Meng S, Shei A, Hodin RA. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci U S A. 1998; 95: 6791-6796.
- Moradei O, Vaisburg A, Martell RE. Histone deacetylase inhibitors in cancer therapy: new compounds and clinical update of benzamide-type agents. Curr Top Med Chem. 2008; 8: 841-858.
- Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB. HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol. 2009; 20: 1293-1302.
- Khan O, La Thangue NB. Drug Insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol. 2008; 5: 714-726.