
Review Article
Austin J Cancer Clin Res 2015;2(2): 1030.
Signaling Pathways in Glioblastoma Cancer Stem Cells: A Role of Stat3 as a Potential Therapeutic Target
Hiroshi Kanno1,2*, Shigeta Miyake1,2 and Satoshi Nakanowatari2
1Department of Neurosurgery, Yokohama City University Graduate School of Medicine, Japan
2Department of Neurosurgery, Yokosuka City Hospital, Japan
*Corresponding author: Hiroshi Kanno, Department of Neurosurgery, Yokohama City University Graduate School of Medicine, 3-9 Fukuura, Kanazawa-ku, Yokohama, 236-0004, Japan.
Received: February 02, 2015; Accepted: March 26,2015; Published: April 03, 2015
Abstract
Glioblastoma cancer stem cells (GCSCs) play an important role in proliferation, invasion, progression, immune evasion, and resistance to radiation in glioblastoma. Their signaling pathways including receptor tyrosine kinase, Akt, MARK, Wnt, Notch, Hedgehog, and JAK/STAT pathways are complicated, but STAT-3 is a convergence point in several important signaling pathways and contributes to the tumor progression by promoting cell proliferation, cell cycle progression, the inhibition of apoptosis, and tissue invasion. Therefore, STAT- 3 is a candidate of therapeutic target of GCSCs. STAT3 is activated through tyrosine phosphorylation by various cytokines and growth factors. STAT-3 is tyrosine phosphorylated by three types of kinases such as receptor tyrosine kinases, JAK family members, and oncogenic kinases including Src and Bcl- Alb. Tyrosine phosphorylated STAT-3 dimerizes and translocates to the nucleus. Active STAT-3 dimers bind to consequences in the promoters of genes such as Bcl-2, Bcl-xL, Mcl-1, and cyclin D1. After induction of target gene expression, multiple STAT-3-endogenous negative regulators such as SOCS3, VHL, and PIAS3 attenuate STAT-3 signaling, and similarly STAT-3 exogenous negative regulators such as pharmacologic JAK inhibitors, dobesilate, and decoy oligonucleotides attenuate STAT-3 activity. In GCSCs, STAT-3 plays a role as a molecular hub in several important signaling pathways that control proliferation, cell cycle progression, anti-apoptosis, invasion, angiogenesis and immune evasion. Therefore, STAT-3 has great potential as a therapeutic target.
Keywords: Signaling pathway; Glioblastoma cancer stem cells; STAT-3; Therapeutic target
Introduction
The cancer stem cells are defined as a cell population residing in tumors, having self -renewal capacity and dividing to give rise to the variety of tumor cells [1,2]. Those cells can reconstitute both the tumor cell hierarchy and the clinical disease state in vivo after xenotransplantation [3]. Although the existence of cancer stem cells in human leukemia is established [1,3], those cells were later discovered to exist in various solid tumors including breast [4], colon [5], lung [6], liver cancers [7], and glioblastoma [8].
Glioblastoma is the most common and lethal brain tumor that shows aggressive natures, with a median overall survival (OS) of less than15 months after diagnosis. The standard first-line treatment includes resection as much as possible, followed by concurrent radioand chemotherapy with temozolomide (TMZ), and then 6–12 months of chemotherapy with TMZ. Despite the treatment, recurrence is universal. Glioblastoma is thought to contain a population of selfrenewing glioblastoma cancer stem cells (GCSCs) that contributes to treatment resistance [9]. GCSCs are functionally defined with selfrenewal measured by serial neurosphere assay in vitro and tumor forming capacity through serial transplantation. GCSCs have been shown to differentiate into astrocytes, oligodendrocytes and neurons [10]. Commonly used GCSC markers are as follows: cell surface: CD133, CD15, CD44, CXCR4; integrin alpha–6; protein (cytoplasmic & nuclear): netin, Musashi-1, Bmi-1; transcriptional factor: Sox2, enzyme, ALDH1 [9]. Recently, researches on glioblastoma have focused on the elucidation of various signaling pathways in GCSCs, particularly aberrant activation. Here, we describe the signaling pathway in GCSCs, particularly focusing on the signal transducer and activator of transcription-3 (STAT-3) as a therapeutic target [10].
Up and downstream signaling pathways of STAT-3 in GSCSs
The activation of several signaling pathways including receptor tyrosine kinase, Akt, MARK, Wnt, Notch, Hedgehog, and JAK/STAT pathways is involved in the progression and proliferation in GCSCs [11]. Among those various signaling pathways in GCSCs, activator of JAK/STAT signal transduction pathway is aberrantly activated in glioblastoma likewise in other solid tumors including breast, lung, ovarian, pancreatic, skin, prostate cancers [12]. STAT-3 contributes to the tumor progression by promoting cell proliferation, cell cycle progression, the inhibition of apoptosis, and tissue invasion [13]. In addition, STAT-3 plays an important role in wound healing, T-cell development, and immune evasion [13,14]. The activation of STAT-3 is implicated in not only controlling critical cellular events involved in tumorigenesis, cell cycle progression, angiogenesis [15], and immune evasion, but also maintaining “stemness” such as self-renewal capacity, ability of differentiation to various tumor cells, and reconstitution of the clinical state in vivo after xeno-transplantation [13,16]. Many tumor-derived cell lines require STAT proteins, particularly STAT-3, to maintain a transformed phenotype [17], and a constitutively active STAT-3 mutant, STAT-3C, was sufficient to transform benign tumor cells to malignant tumor cells, and these transformed cells could form tumors in nude mice [18]. Furthermore, a dominant-negative mutant of STAT-3 blocked transformation by v-src [19]. STAT-3 is activated through phosphorylation of tyrosine 705 that initiates to form a complex composed of two phosphorylated STAT-3 monomers (pSTAT-3). pSTAT-3 homodimers translocate to the nucleus and bind DNA. STAT3 is activated through tyrosine phosphorylation by various cytokines such as interleukin-6 (IL-6) cytokine families (IL-6, oncostatin M, and leukemia inhibitory factor), IL-4, IL-10, IL- 11, and IL-23 and growth factors including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF) and epidermal growth factor (EGF) [20]. In addition, STAT-3 is tyrosine phosphorylated by three types of kinases such as receptor tyrosine kinases including EGFR, FGFR, and PDGFR, Janus kinase (JAK) family members, and oncogenic kinases including Src and Bcl-Alb [21]. Active STAT-3 dimers bind to consequences in the promoters of genes such as Bcl-2, Bcl-xL, Mcl-1, p21WAF1/CIP1, and cyclin D1 [22]. As STAT-3 affects transcription of genes involved in cell cycle and antiapoptosis, regulation of STAT-3 activity is required to prevent malignant transformation of cells. After induction of target gene expression, multiple STAT-3-endogenous negative regulators attenuate STAT-3 signaling. Suppressors of cytokine signaling (SOCS)-3 and von Hipple-Lindau (VHL) proteins down-regulate the upstream kinase activity responsible for STAT-3 phosphorylation [23,24], while the protein inhibitors of activated STAT (PIAS)3 inhibits STAT-3 directly [25]. SOCS-3 and VHL proteins belong to BC-box protein families that contain a BC-box motif corresponding to the binding site of elongin BC [26], while PIAS3 belongs to PIAS families that contain a zinc ring finger domain, an NH2-terminal LXXLL motif, a COOH-terminal acidic domain, a serine/threoninerich domain and PINIT motif involved in the nuclear retention [27]. SOCS-3 and VHL inhibit JAK activation and subsequently attenuate STAT-3 signal transduction in a classic negative feedback loop in the cytoplasm [28], whereas PIAS3 inhibits STAT-3 DNA binding the nucleus and exhibit E3-SUMO (small ubiquitin-like modifier) ligase activity and SUMOylate a variety of transcription factors including p53, c-Jun, and c-Myb [29].
The Notch signaling pathway is involved in cell fate decisions during normal development and in the genesis of glioblastoma [30]. The downstream effects of Notch signaling are highly tissue and time dependent, and Notch has been implicated both in the maintenance of neural progenitors and in the generation of glia during development of the brain [31]. It has been reported previously that in the developing central nervous system, there is cross-talk between the Notch and STAT3 pathways because STAT-3 binds to adjacent site in the Notch1 promoter [32]. Notch pathway genes were up-regulated in GCSCs by constitutive activation of STAT-3 [33], but in contrast the activation and phosphorylation of STAT3 is mediated by the direct binding of several Notch effectors to STAT3 [34].
Inhibiting STAT-3 activity in tumor cells with either dominantnegative or STAT-3 inhibition by STAT-3 siRNA resulted in increased expression of pro-inflammatory mediators including IFN-β, TNF-α, and IL-6 [35]. STAT-3 signaling is dampened by protein tyrosine phosphatases, such as the SH2-domain containing tyrosine phosphatase family (SHP-1, SHP-2), which downregulate STAT-3 activation directly by phosphorylating active STAT-3 complexes [36] (Figure 1).
Inhibitory regulators of STAT-3
STAT-3 is a promising target for GCSCs, not only because it is a convergence point in several important signaling pathways that promote proliferation [15], invasion [37], immune evasion [14,38,39], anti-apoptosis [40], anti-autophagy [41], formation of peritumoral edema [42] and maintenance of “stemness” [15] but also because aberrant STAT-3 activation results from upstream dysregulation [43] (Table 1). There are two approaches to inhibit STAT-3: (1) through exogenous regulators such as RNA interference and chemical inhibitors, and (2) through endogenous regulators such as PIAS3, SOCS-3, and VHL [13] (Table 2). Direct STAT-3 inhibition has been achieved with dominant negative constructs, oligonucleotides, or, phosphopeptidic agents that mimic the native tyrosine 705 containing binding sequence or non-native STAT3-binding sequences [10]. Some platinum compounds interfere with STAT-3 activation and abrogate signaling mediated by constitutively active STAT-3 [44]. Direct inhibition of STAT-3 activity using RNA interference induced apoptosis and inhibited survival [17]. Among decoy oligonucleotides of STAT-3 binding site, G-quartets, having competitive inhibitory structures comprised of guanine-rich oligonucleotides, inhibited STAT-3 binding to endogenous gene promoters and subsequently attenuated STAT-3-induced gene expression by competitively binding activated STAT-3 [45]. STX-0119 (a N-[2-(1,3,4-oxadiazolyl)]-4- quinolinecarboxamide derivative) inhibited STAT3 dimerization and suppressed the growth of transplanted tumors of GCSCs. STX-0119 inhibited proliferation and sphere formation in GCSCs by downregulating the gene expression of STAT3-target genes including cyclin D1, survivin, Bcl-xL, c-myc, MMP2, VEGF and HIF-1α [46]. Similarly, S31-201 inhibited STAT-3 homodimer formation by inhibiting STAT-3 DNA binding, and showed the antitumor activity by inhibiting STAT-3 induced gene expression [47]. In addition, small molecule, nonphosphorylated STAT-3 inhibitor, 31 (SH-4-54) that strongly binds to STAT3 protein. SG-4-54 effectively suppresses STAT3 phosphorylation and its downstream transcriptional target genes and potently kills GCSCs. Moreover, in vivo, SH-4-54 inhibited pSTAT-3 in vivo and potently controlled glioblastoma tumor growth with exhibition of blood-brain barrier permeability [10].
![]()
Authors & Year [Reference]
Role of STAT-3
Hirano T, et al. 2000 [39]
B cell growth and differentiation
Rahaman SO, et al. 2002 [40]
Apoptosis inhibition
Sherry MM, et al. 2009 [16]
Proliferation and maintenance of multipotency in GCSCs
See AP, et al. 2012 [14]
Modulation of immune microenvironment
Yokogami K, et al. 2013 [15]
VEGF upregulation
Zheng Q, et al. 2014 [37]
Tumor invasion
Assi H, et al. 2014 [38]
Dendritic cell differentiation
Wang XF, et al. 2014 [42]
Peritumoral edema formation
Yaun G, et al. 2014 [41]
Autophagy suppression
Table 1: Roles of STAT-3 associated with glioblastoma / glioblastoma cancer stem cells (GCSCs).
![]()
Authors & Year [Reference]
STAT-3 inhibitors
Bromberg JF, et al. 1998 [19]
RNA interference
Dorsey JF, et al. 2000 [52]
PD180970
Leong PL, et al. 2003 [45]
Decoy oligonucleotide
Blaskovich MA, et al. 2003 [50]
JSI-124
Cuevas P, et al. 2005 [54]
Dobesilate
Shuai K. 2006 [25]
PIAS3
Iwamaru A, et al. 2007 [49]
WP1066
Yoshimura A, et al. 2007 [28]
SOCS-3
Iwamaru A, et al. 2007 [49]
AG490
McFarland BC, et al. 2011 [51]
AZD1480
Mir SA, et al. 2012 [47]
S31-201
Kanno H, et al. 2013 [24]
VHL
Ashizawa T, et al. 2013 [46]
STX-0119
Rath KS, et al. 2014 [60]
HO-3867
Kim HS, et al. 2014 [61]
INFPP5F
Table 2: STAT-3 inhibitors.
Inhibition of upstream kinases of STAT-3 leads to abrogation of STAT-3 activity. EGFR inhibitor gefitinib, an upstream kinase of STAT-3, leaded to inhibition of STAT-3 tyrosine phosphorylation [48]. Since the activation of STAT-3 depends on its direct phosphorylation by tyrosine kinases, several pharmacologic JAK inhibitors [AG490, WP1066, JSI-124 (cucurbitacin I), and AZD1480], FGF signaling pathway inhibitor [Dobesilate], and phosphorylation inhibitor [HO-3867] leaded to STAT-3 inhibition [49-56]. The JAK inhibitor AG490 decreased cell survival and induced apoptosis by blocking constitutively active STAT-3 in glioblastoma [49]. AG490 inhibited expression of STAT-3 target- prosurvival genes including Bcl-xL, Bcl-2, and Mcl-1 and leaded to apoptosis [49]. Afterwards the structure of AG490 was modified to produce the more potent and active compound WP1066 that successfully crossed the blood brain barrier and that inhibited the growth of xenografts compared with untreated controls [53]. WP1066 blocked tyrosine phosphorylation of STAT-3 by JAK, which resulted in a failure of STAT-3 to translocate to the nucleus and to mediate its transcriptional effects. WP1066 treatment of glioblastoma cells decreased STAT-3-mediated expression of Bcl-xL, Mcl-1, and c-Myc, induced apoptosis, and reduced viabilities [53]. WP1066 also reversed immune tolerance by blocking STAT-3-mediated immune suppression. WP1066- stimulated proliferation of T cells obtained from glioblastoma patients and enhanced immunogenic responses in glioma-infiltrating microglia, macrophages, and peripheral blood monocytes [53]. Another Jak inhibitor JSI-124 significantly enhanced apoptosis and inhibited proliferation of glioblastoma [49]. Furthermore, it was demonstrated that STAT-3 inhibition by JSI-124 sensitized GBM cells to temozolomide, bis-chloroethylnitrosourea, and cisplatin, enhancing the anti-proliferative effects of these clinically utilized chemotherapeutic agents. Similar additive effects of anti-Stat3 inhibitors with temozolomide or Taxol [50]. In addition, another JAK inhibitor, AZD1480, has been observed to significantly reduce glioblastoma cell growth in both cell culture and animal xenograft studies by blocking STAT-3 activity [51]. Furthermore, Src inhibitor PD180970, a novel pyrido[2,3-d]pyrimidine derivative, reduced STAT3 activity thorough Src inhibition, cell cycle arrest in G2, and reduced viability of cells accompanied by induction of apoptosis [52]. Dobesilate (Dihydroxy-2,5-benzenesulfonate) inhibits activation of STAT-3 by attenuating the upstream of FGF signaling pathway and constitutive expression of tyrosine phosphorylated STAT-3, and blocks neovascularization [54-56], In addition, dobesilate inhibits activation of the MAPK extracellular signal-regulated protein kinases [57] and expression of the prosurvival proteins Bcl-xL and cyclin D1 [58]. These facts suggest that dobesilate increased apoptosis and decreased cell growth and survival partially by blocking STAT- 3 activation. The kinase inhibitors at upstream of STAT-3 have significant potential as chemotherapeutic agents for glioblastoma. Furthermore, inhibition of various upstream kinases in combination with direct STAT-3 inhibitors may also down-regulate GCSCs and be effective in glioblastoma therapy. Recently it has been shown that inhibition of a single tyrosine kinase pathway provides little benefit in reducing glioma cell survival and growth. However, a combinatorial strategy to inhibit multiple upstream tyrosine kinases proved to significantly reduce intracellular signaling, survival, and anchorage-independent growth of glioma cells [58]. Then, STAT- 3 inhibition by direct, indirect, or a combinatorial approach using existing pharmacologic inhibitors has promise as a clinical target in glioblastoma.
HO-3867, a novel curcumin analog and a member of , the diarylidenyl piperidone class decreased expression of tyrosinephosphorylated STAT3 (pTyr705) and STAT3’s downstream targets cyclin D1and Bcl-2, and significantly inhibited cancer stem cell growth in a dose- and time-dependent manner [59,60]. INPP5F (inositol polyphosphate-5-phosphatase F), one of the polyphosphoinositide differentially expressed in GCSCs, inhibited STAT-3 activity via inhibition of STAT-3 phosphorylation and suppressed glioma tumorigenesity [60]. Constitutively expressed INPP5F showed to suppress self-renewal and proliferation potentials of glioblastoma cells and reduced tumorigenicity of glioblastoma. In addition, loss of INPP5F gene in gliomas is significantly correlated with lower overall patient survivals. INPP5F is a potential tumor suppressor in gliomas via endogenous inhibition of STAT3 pathway [61].
SOCS-3 and VHL are not only endogenous inhibitors in STAT- 3 signaling [24,62] and they work in a negative-feedback loop to suppress STAT-3 signaling [63,64]. In contrast, elevated SOCS-3 or VHL expression was reported in some cancer tissues [65-70]. It was shown that glioblastoma tissues expressed higher levels of SOCS-3 than did control brain tissues and that SOCS-3 promoter activity and mRNA expression is inhibited by ectopic PIAS3 expression [71]. Moreover, constitutive expression of SOCS-3 in glioblastoma is related to cell proliferation and resistance to radiation [72]. U87-MG glioma cells constitutively express SOCS-3 [64], while both those cells and GCSCs derived from U87-MG glioma do not express VHL [24]. A unique characteristic of GCSCs is the expression of the cell surface marker CD133 that is a hallmark of neural precursor cells [73]. Since the expression of SOCS-3 in glioblastoma cells was shown to correlate with resistance to ionizing radiation [71,72], CD133+ cells might also express elevated levels of SOCS-3 as well as activated STAT-3.
Similarly, VHL inhibited JAK2 and STAT3. In addition, VHL upregulated PTEN expression. This fact suggested that VHL affected the JAK/STAT pathway as well as the PTEN/PI3K/Akt pathway; they also suggest that up-regulation of PTEN by VHL gene transfer may affect the PI3K/Akt pathway, since PTEN is a PI3K/Akt pathway inhibitor [73]. VHL inhibited the implantation ability as well as soft agar colony and neurosphere formation, all of which are characteristics of cancer stem cells. Our results suggest that these characteristics may be related to the JAK/STAT or PTEN/PI3K/Akt pathways in GCSCs. Overexpression of VHL up-regulated PTEN and down-regulated the JAK/STAT pathway. In addition, overexpression of VHL significantly down-regulated the proliferation of U87-GCSCs. However, the overexpression of VHL inhibited cell proliferation of U87-GCSCs to a lesser extent than it did soft agar colony and neurosphere formation, or implantation capacity into SCID mice. These results suggest that VHL inhibited the tumorigenicity and self-renewal ability via the JAK/STAT pathway and also affected the PTEN/PI3K/Akt pathway, both of which are critical in GCSCs [24] (Figure 1).
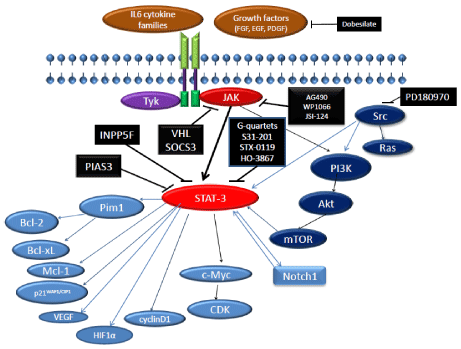
Figure 1: Signaling pathways in GCSCs. The binding of cytokine and growth
factor to their corresponding receptors leads to downstream activation of
STAT-3 either directly or through intermediate substrates such as JAK, Src,
and Trk. STAT-3 enters the nucleus and induces many gene expressions
involved in cell cycle progression, anti-apoptosis, angiogenesis, migration,
and invasion. STAT-3 activity is down-regulated by INPP5F, SOCS3, VHL
and PIAS3. In addition, several inhibitors targeting the JAK and Src downregulate
STAT-3, and there is a cross talk between Notch and STAT-3.
Conclusion
In GCSCs, STAT-3 plays a role of a molecular hub in several important signaling pathways that control proliferation, cell cycle progression, anti-apoptosis, invasion, angiogenesis and immune evasion. There are two approaches to inhibit STAT-3: (1) through endogenous inhibitors (PIAS3, SOCS-3, VHL) and (2) through exogenous inhibitors (JAK inhibitors, Src inhibitor, EGFR inhibitor, decoy oligonucleotides, RNA interface). STAT-3 has great potential as a therapeutic target.
References
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3: 730-737.
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414: 105-111.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367: 645-648.
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003; 100: 3983-3988.
- Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol Med (Berl). 2009; 87: 1097-1104.
- Li N, Yang Y, Ding M, Huang W, Li H, Ye J, et al. GFP stable transfection facilitated the characterization of lung cancer stem cells. Mol Biotechnol. 2014; 56: 1079-1088.
- Pan QZ, Pan K, Wang QJ, Weng DS, Zhao JJ, Zheng HX, et al. Annexin A3 as a potential target for immunotherapy of liver cancer stem-like cells. Stem Cells. 2015; 33: 354-366.
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003; 63: 5821–5828.
- Khan IS, Ehtesham M. Targeting glioblastoma cancer stem cells: the next great hope? Neurosurg Focus. 2014; 37: E7.
- Haftchenary S, Luchman HA, Jouk AO, Veloso AJ, Page BD, Cheng XR, et al. Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med Chem Lett. 2013; 4: 1102-1107.
- Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol. 2010; 80: 654-665.
- Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014; 1845: 136-154.
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res. 2008; 6: 675-684.
- See AP, Han JE, Phallen J, Binder Z, Gallia G, Pan F, et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J Neurooncol. 2012; 110: 359-368.
- Yokogami K, Yamashita S, Takeshima H. Hypoxia-induced decreases in SOCS3 increase STAT3 activation and upregulate VEGF gene expression. Brain Tumor Pathol. 2013; 30: 135-143.
- Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009; 27: 2383-2392.
- Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002; 109: 1139-1142.
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999; 98: 295-303.
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE Jr. Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998; 18: 2553-2558.
- Luwor RB, Stylli SS, Kaye AH. The role of Stat3 in glioblastoma multiforme. J Clin Neurosci. 2013; 20: 907-911.
- Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007; 13: 5665-5669.
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000; 19: 2474- 2488.
- Ramana CV, Kumar A, Enelow R. Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2005; 327: 727-733.
- Kanno H, Sato H, Yokoyama TA, Yoshizumi T, Yamada S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int J Oncol. 2013; 42: 881-886.
- Shuai K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006; 16: 196-202.
- Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004; 18: 3055-3065.
- Duval D, Duval G, Kedinger C, Poch O, Boeuf H. The 'PINIT' motif, of a newly identified conserved domain of the PIAS protein family, is essential for nuclear retention of PIAS3L. FEBS Lett. 2003; 554: 111-118.
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007; 7: 454-465.
- Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc Natl Acad Sci U S A. 2002; 99: 2872-2877.
- Stockhausen MT, Kristoffersen K, Poulsen HS. The functional role of Notch signaling in human gliomas. Neuro Oncol. 2010; 12: 199-211.
- Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002; 25: 471-490.
- Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor κB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem. 2013; 288: 26167-26176.
- Zhang X, Zhao X, Shao S, Zuo X, Ning Q, Luo M, et al. Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int J Oncol. 2015; 46: 1141-1148.
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004; 10: 48-54.
- Rakesh K, Agrawal DK. Controlling cytokine signaling by constitutive inhibitors. Biochem Pharmacol. 2005; 70: 649-657.
- Villalva C, Martin-Lannerée S, Cortes U, Dkhissi F, Wager M, Le Corf A, et al. STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer. 2011; 128: 826-838.
- Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. 2014, 16: 1229-1243.
- Assi H, Espinosa J, Suprise S, Sofroniew M, Doherty R, Zamler D, et al. Assessing the role of STAT3 in DC differentiation and autologous DC immunotherapy in mouse models of GBM. PLoS One. 2014; 9: e96318.
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000; 19: 2548-2556.
- Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002; 21: 8404-8413.
- Yuan G, Yan SF, Xue H, Zhang P, Sun JT, Li G, et al. Cucurbitacin I induces protective autophagy in glioblastoma in vitro and in vivo. J Biol Chem. 2014; 289: 10607-10619.
- Wang XF, Lin GS, Lin ZX, Chen YP, Chen Y, Zhang JD, et al. Association of pSTAT3-VEGF signaling pathway with peritumoral edema in newly diagnosed glioblastoma: an immunohistochemical study. Int J Clin Exp Pathol. 2014; 7: 6133-6140.
- 43. Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004; 3: 1533-1542.
- Jing N, Li Y, Xu X, Sha W, Li P, Feng L, et al. Targeting Stat3 with G-quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003; 22: 685-696.
- Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci U S A. 2003; 100: 4138-4143.
- Ashizawa T, Miyata H, Iizuka A, Komiyama M, Oshita C, Kume A, et al. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int J Oncol. 2013; 43: 219-227.
- Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S, et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem. 2012; 287: 2666-2677.
- Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000; 6: 2053 –2063.
- Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene. 2007; 26: 2435-2444.
- Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003; 63: 1270– 1279.
- McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011; 10: 2384-2393.
- Dorsey JF, Jove R, Kraker AJ, Wu J. The pyrido[2,3-d]pyrimidine derivative PD180970 inhibits p210Bcr-Abl tyrosine kinase and induces apoptosis of K562 leukemic cells. Cancer Res. 2000; 60: 3127-3131.
- Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007; 67: 9630-9636.
- Cuevas P, Sanchez I, Lozano RM, Gimenez-Gallego G. Dobesilate is an angiogenesis inhibitor. Eur J Med Res. 2005; 10: 369-372.
- Tierney BJ, McCann GA, Cohn DE, Eisenhauer E, Sudhakar M, Kuppusamy P, et al. HO-3867, a STAT3 inhibitor induces apoptosis by inactivation of STAT3 activity in BRCA1-mutated ovarian cancer cells. Cancer Biol Ther. 2012; 13: 766-775.
- Cuevas P, Díaz-González D, Giménez-Gallego G, Dujovny M. Dihydroxy-2,5 benzenesulphonate (dobesilate) elicits growth arrest and apoptosis in glioma cells. Neurol Res. 2005; 27: 797-800.
- Cuevas P, Díaz-González D, Sánchez I, Lozano RM, Giménez-Gallego G, Dujovny M, et al. Dobesilate inhibits the activation of signal transducer and activator of transcription 3, and the expression of cyclin D1 and bcl-XL in glioma cells. Neurol Res. 2006; 28: 127-130.
- Cuevas P, Díaz-González D, García-Martín-Córdova C, Sánchez I, Lozano RM, Giménez-Gallego G, et al. Dobesilate diminishes activation of the mitogen-activated protein kinase ERK1/2 in glioma cells. J Cell Mol Med. 2006; 10: 225-230.
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007; 318: 287-290.
- Rath KS, Naidu SK, Lata P, Bid HK, Rivera BK, McCann GA, et al. HO-3867, a safe STAT3 inhibitor, is selectively cytotoxic to ovarian cancer. Cancer Res. 2014; 74: 2316-2327.
- Kim HS, Li A, Ahn S, Song H, Zhang W. Inositol Polyphosphate-5-Phosphatase F (INPP5F) inhibits STAT3 activity and suppresses gliomas tumorigenicity. Sci Rep. 2014; 4: 7330.
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007; 7: 454-465.
- Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997; 387: 917-921.
- Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN, et al. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007; 179: 5966-5976.
- Brender C, Lovato P, Sommer VH, Woetmann A, Mathiesen AM, Geisler C, et al. Constitutive SOCS-3 expression protects T-cell lymphoma against growth inhibition by IFNalpha. Leukemia. 2005; 19: 209-213.
- Evans MK, Yu CR, Lohani A, Mahdi RM, Liu X, Trzeciak AR, et al. Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene. 2007; 26: 1941-1948.
- Raccurt M, Tam SP, Lau P, Mertani HC, Lambert A, Garcia-Caballero T, et al. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer. 2003; 89: 524-532.
- Zhang S, Zhou X, Wang B, Zhang K, Liu S, Yue K, et al. Loss of VHL expression contributes to epithelial-mesenchymal transition in oral squamous cell carcinoma. Oral Oncol. 2014; 50: 809-817.
- Ferchichi I, Kourda N, Sassi S, Romdhane KB, Balatgi S, Cremet JY, et al. Aurora A overexpression and pVHL reduced expression are correlated with a bad kidney cancer prognosis. Dis Markers. 2012; 33: 333-340.
- Corless CL, Kibel AS, Iliopoulos O, Kaelin WG Jr. Immunostaining of the von Hippel-Lindau gene product in normal and neoplastic human tissues. Hum Pathol. 1997; 28: 459-464.
- Zhou H, Miki R, Eeva M, Fike FM, Seligson D, Yang L, et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res. 2007; 13: 2344-2353.
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006; 444: 756-760.
- Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009; 4: 226-235.