
Case Report
Austin J Cancer Clin Res 2015;2(5): 1046.
Isolated Skull Metastasis of Ewing’s Sarcoma in a Child
Puerta P¹*, Guillén A¹, Mora J², Suñol M³ and Ferrer E¹
¹Department of Neurosurgery, University of Barcelona, Spain
²Department of Oncology, University of Barcelona, Spain
³Department of Pathology, University of Barcelona, Spain
*Corresponding author: Puerta P, Department of Neurosurgery, University of Barcelona, Passeig Sant Joan de Déu 2, 08950, Spain.
Received: May 04, 2015; Accepted: June 09, 2015; Published: July 30, 2015
Abstract
Ewing’s sarcoma (ES) is a rare primary malignant bone tumor usually occurring in children and young adults. It most often affects long bones and pelvis. Metastatic lesions usually occur in lung, bone and the bone marrow. Central nervous system involvement is rare and it most frequently presents with other evidence of tumor recurrence. We present a case of isolated metastasis of ES involving the parietal bone in a 10-year old boy. To our knowledge, three other cases of skull metastasis of ES have been published previously. All of them presented other systemic metastases at the same time. The surgical indication for skull metastasis of ES depends on its location and general condition. Resection of solitary skull metastasis of ES should be as radical as possible, followed by systemic chemotherapy.
Keywords: Ewing’s sarcoma; Skull metastasis; CNS involvement; Children
Case Presentation
Ewing’s sarcoma (ES) was first described by James Ewing in 1921 as a “diffuse hemangioendothelioma of bone” [1,2]. It is a tumor of childhood that arises from the neuroectodermal cells of the medullary cavity [2,3]. ES is a relatively rare tumor in neurosurgical practice. It usually affects long bones of the lower limbs and pelvis [1,2,4,-6]. The predominant sites of metastases include lung, bone and the bone marrow [7]. Metastasis of ES to the central nervous system (CNS) is uncommon [7-9] and skull metastasis of ES in children is extremely rare [6]. We report a 10-year-old patient with an isolated parietal bone metastasis of ES.
A 10-year-old male underwent resection of an ES in the left 5th rib 15 months previously, followed by chemotherapy and local radiation treatment. He had a painless, progressive swelling over his right parietal area. There was no history of headache, vomiting or seizures. Neurological examinations revealed no findings. Physical examination revealed a mass of 4cm in diameter over the right parietal region which was circumscribed, hard and nonpulsatile.
Computed tomography (CT) scan showed an isodense parietal tumor which was homogeneously enhanced by contrast medium. It was partly extracranial and partly intracranial, from de epidural space to the scalp with bone destruction (Figure 1a, b). The FDG PET scan only demonstrated increased of metabolism in the parietal mass, suggesting that it was an isolated metastasis. Bone marrow biopsies were negative.
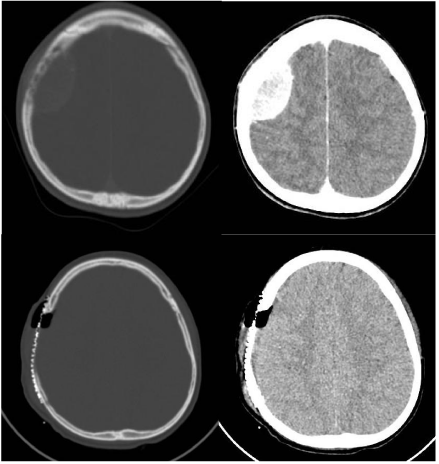
Figure 1: Preoperative and postoperative CT scan. 1a,b: Axial-CT scan at
admission shows an intracranial extradural lytic lesion of the parietal bone.
1c,d: Postoperative axial CT scan shows no residual tumour and a titanium
mesh cranioplasty.
Gross total removal of the tumor was performed via a right parietal craniectomy and the infiltrated dura was excised (Figure 2). We performed a duraplasty using polyester urethane graft and a cranioplasty using titanium mesh.
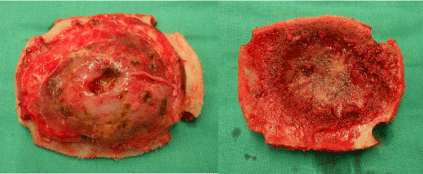
Figure 2: Operative photographs showing the parietal calvarial tumour. a.
Exocraneal side. b. Endocraneal side.
A control CT scan performed within 24-h post-surgery showed total excision of the tumor and postoperative changes (Figure 1c, d).
Histopathology showed an extensive intraosseous tumoral infiltration composed of uniform small round cells, with involvement of subcutaneous soft tissue. The dura was also infiltrated by sheets and nests of tumoral cells. The tumoral cells showed strong membranous CD99 staining (Figure 3). The histology was identical to the specimens from the left rib tumor. The diagnosis was skull metastasis of ES.
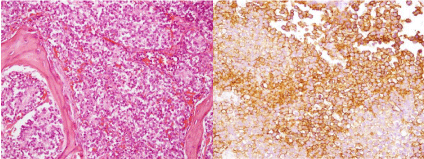
Figure 3: Histopathology. a. Sheets of uniform small round cells. b. Strong
membranous CD99 staining.
Based on the malignant nature of the tumor and after complete macroscopic resection, postoperative systemic chemotherapy with irinotecan-temozolamide and local radiotherapy were performed. He is still alive and no tumor recurrence has presented in the 8-months post surgery follow-up.
Discussion
ES is the second most common malignant bone tumor [1]. It usually occurs in patients younger than 20 years of age and the peak incidence is observed in 5-13-year –old patients. ES is more frequent in males than females [1,10].
Primary ES most often originates in long bone shafts (47%), pelvic bones (29%), ribs and vertebrae (12%) [1]. This tumor frequently develops metastases. In 75-80% of cases, the metastases manifest themselves during the first two years after primary tumor had been diagnosed. The principal sites of metastases are lung, bone and the bone marrow [1,7].
Metastasis to the CNS has been reported in 10-37% of cases [1,6,9,11]. Meningeal invasion and spinal cord compression are the best known forms of CNS involvement [7,9,12]. Intraparenchymal metastases of ES are also reported [12,13].
The frequency of skull metastases of ES in children is unknown. To our knowledge, there are only three cases of metastatic ES of the skull reported in children, all of them at relapsed [6]. Summary data of these patients are shown in Table 1. Hattori et al. treated three patients with skull metastasis of ES. All their three cases occurred on the midline of the skull, just above the superior sagittal sinus. This fact can be explained because the midline of the skull is the watershed area of the external carotid artery resulting in harboring of metastatic tumor cells [6]. However, our patient presented a parietal tumor in the right side, without involvement of the midline or superior sagittal sinus.
![]()
Case
Location of primary tumor
Time of skull metastasis
Additional sites of metastases
Follow-up
1 (Hattori et al)
Mandibula
At relapse
Femur
Alive 11-year postsurgery
2 (Hattori et al)
Rib
At relapse
Scapula
Alive 1-year postsurgery
3 (Hattori et al)
Femur
At relapse
Ribs, vertebrae
Dead 4-months postdiagnosis
4 (Present case)
Rib
At relapse
-
Alive 8 months post-surgery
Table 1: Case summary of patients with skull metastasis of ES.
Isolated brain or meningeal disease is uncommon (2, 2%). CNS disease most frequently occurs with other evidence of tumor recurrence [12,14]. In fact, all three cases of skull metastases of ES reported by Hattori et al. presented other systemic metastases at the same time. Our patient had an isolated skull metastasis of ES 15 month’s later diagnosis of his disease.
It has been reported that skull metastasis of ES is not rare compared to primary ES of the skull [6]. Dahlin and Unni reported that Ewing’s sarcoma is quite uncommon as a primary skull lesion, occurring in 1% of cases [15]. However, there are many more cases of primary ES of the skull than metastatic lesions published in the English literature [2,4,9,11].
The multimodal treatment for ES is currently considered to be the optimal treatment method. It includes the maximum resection of the tumor followed by radiation and chemotherapy [1,16]. Tumor resection should be as radical as possible within the admissible safety limits [1,7]. Combined modality treatment is also accepted to be an essential requirement in relapsed ES. Local therapy of relapse is associated with better prognosis, including surgery and/or radiotherapy. Despite there is no standardized second line treatment for relapsed ES, combined modality treatment of the metastases must complement systemic chemotherapy whenever possible [17]. Solitary skull metastasis should be removed totally, followed by systemic chemotherapy. Aggressive treatment should be considered for the metastatic lesion depending on other systemic metastases and the general condition [6].
The materials used to performed cranioplasties after skull tumor resections have been autologous iliac crest, calvarial bone graft and homologous bone grafts. In complex skull defects or children who need MRI control, the use of custom-made prostheses should be considered [18]. However, we performed a cranioplasty with titanium mesh because the size and location of the skull bone defect.
The prognosis for patients with ES used to be unfavourable. In 1974, Rosen implemented the combination of postoperative multimodal chemotherapy and radiation therapy, so the 5-year survival rate increased from 10 to 55-60% [1,2,11]. The presence of distant metastases by the time of diagnosis is the most unfavourable prognostic factor in patients with ES [1]. About 30-40% of patients with ES who are initially diagnosed with localized primary and 60- 80% of those with primary disseminated disease do relapse. Prognosis of patients with relapsed ES is poor. Relapses within 24 months of initial diagnosis expect a worse prognosis and the 5-year survival rate decreased to 7% [17].
We present a case of isolated skull metastasis of ES involving the parietal bone in a 10-year-old boy. Three other cases of metastatic ES of the skull have been reported previously in children. Prognosis of these patients is poor but local therapy of relapsed ES is associated with a more favourable outcome. Aggressive multimodal treatment is advisable in relapsed ES. So, systemic chemotherapy should be considered after local treatment.
References
- Cherekaev VA, Kushel' IuV, Shkarubo AN, Mukhametzhanov DZh, Stepanian MA, Rotin DL. [Primary and metastatic Ewing sarcoma of the skull base - case reports and comparative analysis]. Zh Vopr Neirokhir Im N N Burdenko. 2013; 77: 30-36.
- Ahmad FU, Suri A, Mahapatra AK, Ralte A, Sarkar C, Garg A. Giant calvarial Ewing's sarcoma. Pediatr Neurosurg. 2004; 40: 44-46.
- Cavazzana AO, Miser JS, Jefferson J, Triche TJ. Experimental evidence for a neural origin of Ewing's sarcoma of bone. Am J Pathol. 1987; 127: 507-518.
- Carlotti CG Jr, Drake JM, Hladky JP, Teshima I, Becker LE, Rutka JT. Primary Ewing's sarcoma of the skull in children. Utility of molecular diagnostics, surgery and adjuvant therapies. Pediatr Neurosurg. 1999; 31: 307-315.
- Bhansali SK, Desai PB. Ewing’s sarcoma: observations on 107 cases. J Bone Joint Surg. 1963; 45: 541-553.
- Hattori T, Yamakawa H, Nakayama N, Kuroda T, Andoh T, Sakai N, et al. Skull metastasis of Ewing's sarcoma--three case reports. Neurol Med Chir (Tokyo). 1999; 39: 946-949.
- Kim EY, Lee SK, Kim DJ, Kim J, Lee KS, Jung W, et al. Intracranial dural metastasis of Ewing's sarcoma: a case report. Korean J Radiol. 2008; 9: 76-79.
- Postovsky S, Ash S, Ramu IN, Yaniv Y, Zaizov R, Futerman B, et al. Central nervous system involvement in children with sarcoma. Oncology. 2003; 65: 118-124.
- Colak A, Berker M, Ozcan OE, Erbengi A. CNS involvement in Ewing's sarcoma. A report of 12 cases. Acta Neurochir (Wien). 1991; 113: 48-51.
- Falk S, Alpert M. Five-year survival of patients with Ewing's sarcoma. Surg Gynecol Obstet. 1967; 124: 319-324.
- Desai KI, Nadkarni TD, Goel A, Muzumdar DP, Naresh KN, Nair CN. Primary Ewing's sarcoma of the cranium. Neurosurgery. 2000; 46: 62-68.
- Trigg ME, Glaubiger D, Nesbit ME Jr. The frequency of isolated CNS involvement in Ewing's sarcoma. Cancer. 1982; 49: 2404-2409.
- Marciani MG, Stefani N, Peroni L, Stefanini F, Tarantino U, Gigli GL, et al. Intracerebral metastasis in Ewing's sarcoma. Acta Neurol Belg. 1990; 90: 149-156.
- Kies MS, Kennedy PS. Central nervous system involvement in Ewing's sarcoma. Ann Intern Med. 1978; 89: 226-227.
- Dahlin DC, Unni KK. Bone tumours. General aspects and data on 8542 cases. Ch C Thomas, Springfield. 1986; III: 332-336.
- Kadar AA, Hearst MJ, Collins MH, Mangano FT, Samy RN. Ewing's Sarcoma of the Petrous Temporal Bone: Case Report and Literature Review. Skull Base. 2010; 20: 213-217.
- Rasper M, Jabar S, Ranft A, Jürgens H, Amler S, Dirksen U. The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer. 2014; 61: 1382-1386.
- Castle M, Rivero M, Marquez J. Primary Ewing's sarcoma of the skull: radical resection and immediate cranioplasty after chemotherapy. A technical note. Childs Nerv Syst. 2013; 29: 303-306.