
Special Article - Advanced Prostate Cancer
Austin J Cancer Clin Res. 2017; 4(1): 1074.
5α- Reductase Inhibitors in Prostate Cancer: An Overview
Neelima Dhingra*
Department of Pharmaceutical Chemistry, Panjab University, India
*Corresponding author: Neelima D Passi, Department of Pharmaceutical Chemistry, University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh- 160014, India
Received: February 28, 2017; Accepted: April 10, 2017; Published: April 25, 2017
Abstract
Prostate cancer is the noncutaneous malignancy of the prostate gland in elderly men. Steroid 5α-reductase (5AR) enzyme dictates the cellular availability of dihydrotestosterone to prostatic epithelial cells and consequently modulates its growth. Excessive production of DHT has been implicated in the pathogenesis of Prostate cancer. Finasteride and Dutasteride, clinically available 5AR inhibitors may play an important role in the prevention and treatment of prostate cancer by blocking peripheral conversion of T into DHT. This article gives a brief account of biology of prostate, rationale and efficacy of 5AR inhibitors in prostate cancer management and their associated controversies.
Keywords: Prostate; Androgens; Finasteride; Dutasteride
Introduction
Prostate cancer (PC) is the noncutaneous malignancy and second leading cause of cancer death in American men [1]. It is the most commonly disease of elderly age men and the average age at the diagnosis is 66. The prevalence of prostate cancer increases with increasing age and 27.3% of all new diagnoses are in men 75 years of age or older [2]. Depending on the severity or stage, symptoms may vary from urinating problems, including a slow or weak urinary stream or the need to urinate more often, especially at night; blood in the urine or semen; erectile dysfunction; pain in the hips, back, chest or other areas from cancer that has spread to bones; weakness or numbness in the legs or feet, or even loss of bladder or bowel control from cancer pressing on the spinal cord [3]. Some time urinating problems are much more often caused by closely related a noncancerous growth of the prostate benign prostatic hyperplasia (BPH).
Prostate cancer is a heterogeneous and biologically complex disease, as a result of which it poses challenges in screening, diagnosis, and disease characterization. Recently the characteristics of prostate cancer have changed dramatically by the introduction of prostatespecific antigen (PSA)-based screening in 1986 [4]. PSA screening has led to a drastic increase in the detection rate of prostate cancer along with an associated downward stage migration.
Closely related medical abnormality in men with prostate cancer is High-grade prostatic intraepithelial neoplasia (HGPIN) that shares clinical, morphological, genetical and molecular signatures [5]. HGPIN has been established as a pre-cursor to prostatic adenocarcinoma, as it tends to occur in the peripheral zone of the prostate (a area where most cases of prostate cancer develop) and an autopsy studies has shown that 82% of prostate cancer specimens also had area of HGPIN, while only 43% of those without prostate cancer. In addition like prostate cancer, with age it becomes increasingly multifocal and the estimated time frame to disease progression after HGPIN findings has been reported to be between 29 and 36 months. It has become clinically important finding on prostate biopsy in terms of possessing high predictive value for future adenocarcinoma cancer, but prostate cancer investigators need to be aware of the potentially overlapping genotype and phenotype between HGPIN and prostatic adenocarcinoma because of the implications upon experimental design and data interpretation [6]. Recently, its predictive value for the development of cancer during the prostate-specific antigen screening era has decreased, mostly owing to the increase in prostate biopsy cores.
The high prevalence and considerably lower mortality of prostate cancer, coupled with the significant potential morbidity of therapy for prostate cancer, have sparked much interest in alternative approaches against prostate cancer viz; active surveillance strategies, surgical therapies and chemotherapy [7-9]. Though the development and overgrowth of the prostate has been attributed to the number of the different factors like aging, late activation of cell growth by mutations in oncogenes, defective or mutated tumors suppressor gene, genetic factors and hormonal changes [3]. But overabundance of dihydrotestosterone (DHT) has been implicated in the pathogenesis of benign prostatic hyperplasia (BPH) and untreated complications prostate cancer [10]. Preventing DHT synthesis via 5α-reductase (5-AR) inhibition has been shown to have a remarkable effect on prostatic disease with low toxicity. Thus, there is much interest in the potential role for 5-AR inhibitors (5-ARIs) in the management of prostatic intraepithelial neoplasia (PIN) and the prevention of prostate cancer. This report reviews knowledge about the role of androgens and the 5-AR system in prostate disease, pharmacology of 5AR inhibitors and highlights their role in prevention and treatment of prostate cancer along with some controversies.
Prostate: an androgen dependent organ
The prostate an important organ of male reproductive system is located between the bladder and the rectum. Walnut sized prostate gland is a heterogeneous organ consisting of central peripheral and transition zone and is composed of three different types of cells: glandular epithelial cells, smooth muscle cells and stromal cells (Figure 1) [11]. The physiologic functions and pathologic conditions of the prostate, like other glands, are regulated by endogenous hormones (androgens i.e. testosterone and dihydrotestosterone) and growth factors. Androgen production is controlled by the hypothalamus and the pituitary gland (Figure 2). More than 98% of all testosterone (T) in the prostate is of testicular origin and others 5-10% is being produced by adrenal gland. (T) is a major androgen in adult male and its bioavailability is directly linked to the prostate development, differentiation. Serum T level rises dramatically in males between the 10-20 years of age and pronounced exponential growth of the prostate is controlled by the balanced agonist and antagonist abilities of androgens to stimulate cell proliferation and to inhibit the rate of cell death in prostate tissue. After the age of 20 years, and under the continuing presence of T, the healthy prostate achieves a steady state of self renewal and maintenance [12,13].
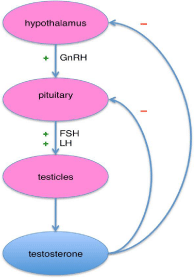
Figure 1: Androgen release.
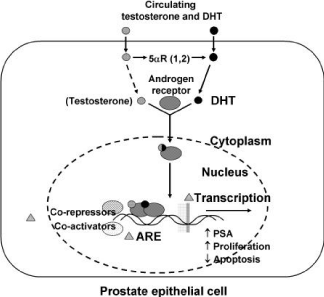
Figure 2: Conversion of Testosterone into dihydrotestosterone in prostatic
epithelial cell.
Within the prostate, the unbound T diffuses into prostate cell, and is converted to dihydrotestosterone (DHT) under the catalytic effect of NADPH dependent enzyme 5-reductase. In the nucleus, DHT on binding with cytosol androgen receptor protein (AR) stimulates the RNA synthesis after interacting with DNA binding sites (Figure 3) [14]. Serum T concentration is approximately 10 times higher than DHT, but in the prostate gland, the ratio is more or less reverse with lesser binding affinity to the androgen receptor than that of DHT. The physiological role of T and DHT is quite different. In the embryo, T stimulates the transformation of Wollffian ducts in epididymis, differential ducts and seminal vesicle and activates the expression of 5α-reductase with the subsequent production of DHT. Whereas, DHT is determinant for the sexual differentiation of male foetus organ with the formation of external genitalia, urethra and prostate in the embryo. After the puberty, T determines the modification of external genitalia, increase of muscle mass, deeping of voice, spermatogenesis, sexual potency and male sexual behavior in males and the increase of body hair, facial hair and the enlargement of prostate is related with DHT formation. Thus DHT remains at high levels in the prostate throughout life, without the age related decline seen in circulating testosterone. It has been well postulated by Burckovsky and Wilson that testosterone acted as prohormone and dihydrotestosterone was the main active hormone in androgen sensitivity [15].
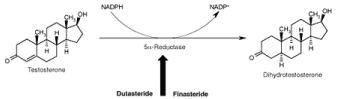
Figure 3: Mechanism of Finasteride and Dutasteride.
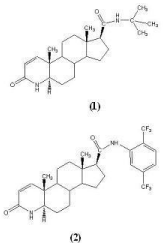
Figure 4: Structures of Finasteride (1) and Dutasteride (2).
Androgen deprivation therapy in the treatment of prostate cancer
Prostatic diseases represent some of the greatest threats to men’s health and the heterogeneous behavior of prostate cancer as well as multiple factors limiting patient accrual are some of the inherent challenges in the development and completion of clinical trials comparing various available therapeutic modalities ranging from active surveillance, surgical extirpation, surgical ablation, radiation therapy, hormonal therapy, to chemotherapy [7-9]. The specific approach used to treat PC depends upon number of factors like age, prostrate size, weight, prostate specific antigen level and stage. As long as symptoms are mild, without histological evidence for bladder or urinary tract deterioration, watchful waiting is deemed appropriate [3]. While surgical extirpation or radiation therapy are considered for the treatment of localized prostate cancer, and hormonal therapy for locally advanced disease, biochemical recurrence after localized therapy, and metastatic disease [8].
Androgen has been found to play a pivotal role in the control of prostatic growth and ablation of their action has been pursued as therapeutic option in PC treatment. Biological basis of androgen ablation therapy lies in the observation that the embryonic development of the prostate is dependent on the androgen dihydrotestosterone and furthermore, castration in men prior to puberty resulted into regression of prostatic enlargement [16]. Reversible androgen deprivation can be achieved by the use of progestational agents [17], gonadotropin releasing hormone (GnRH) analogues [18], antiandrogen [19] and aromatase inhibitors [20] by blocking the peripheral conversion. However, these centrally acting drug got limited clinical efficacy and are poorly tolerated, with a high percentage of patients experiencing erectile dysfunction, loss of libido, “hot flashes”, osteoporosis, fatigue, and muscle wasting [21,22]. Associated side effects and non specific action of all above categories led to the investigation of new and more specific approach 5α-reductase inhibitors.
5α-Reductase inhibitors
5α-Reductase is a NADPH dependent membrane bound enzyme responsible for the conversion of testicular T into DHT, thus dictates its cellular availability to prostatic epithelial cells and consequently modulate prostate growth [23]. Two isozymes of 5α-reductase (5α-reductase type 1 and 5α-reductase type 2) have been cloned, expressed and characterized based on differences in their chromosomal localization, tissue expression pattern and biochemical properties [24]. The type 2 isozyme has been found to be predominant in the prostate, genital skin, seminal vesicle, epididymis and liver and essential for differentiation of male external genitalia during foetal life, as its deficiency in gene leads to the male pseudohermaphroditism. The type 1 is not the major species expressed in the prostate and exhibit micromolar affinities for steroidal substrate i.e. testosterone. Two isoforms possess optimal enzymatic activity at pH-8.5 and pH 4.7- 5.5 respectively [23]. Further, chemical and kinetic mechanisms of 5α-reductase for the irreversible conversion of 4-en-3-oxo-steroid (T) to the corresponding 5α-H-3-oxo-steroid (DHT) conversion have been investigated and found to involves the formation of binary complex between enzyme and NADPH, followed by formation of ternary complex with substrate testosterone [25,26].
The control of the biological action of single steroid DHT, through the inhibition of specific enzyme 5AR involved in its synthesis and metabolism, without significant change in the overall profile of other hormone (T) has been a major theme in the design of 5ARIs. The identification of two isozymes of 5α-reductase, their relative role in physiological and pathological developments of prostatic diseases has opened the door to synthesize more specific and selective inhibitors. Hundreds of steroidal and non-steroidal inhibitors ranging from classical, reversible and irreversible inhibitors, and transition state analogues to mechanism-based analogues have been synthesized during last two decades [27,28]. These agents suppress the DHT concentration by blocking the enzyme, resulting in shrinkage in the size of prostate, increased peak urinary flow rates and ultimately providing relief [29]. Further, the rationale for use of 5ARIs is rooted in the observation that these are more specific to DHT androgens action without affecting/lowering testosterone level, thus capable of decreasing long term side effect of castration due to loss of testosterone without compromising the efficacy of hormonal therapy [30].
Finasteride: Finasteride (MK-906) synthesized in 1984, is chemically 17β-(N-tert-butyl-carbamoyl)-4-aza-5α-androst-1-en-3- one and was the first clinically approved 5ARI in U.S. in 1992 [31]. It is a competitive inhibitor of 5α-reductase type 2 with 10 fold high affinity than type 1 and forms a stable complex with enzyme (Figure 4). It has been reported that at clinical doses of 5 mg/day in human, it decreases the prostate DHT level by 70-90%, thus resulting in decreased prostate volume or size and improved urinary flow rate in BPH patients [32]. It has neither androgenic, antiandrogenic, other hormone related properties, nor it interferes with the binding of T or DHT to the AR [33]. It is being reported that androgen deprivation therapy, with finasteride, induce distinctive histological changes in benign and neoplastic prostatic epithelial cells. Decreased level of DHT, a very small prostate, and complete lack of prostatic glandular epithelium in men with congenital deficiency of 5AR, as well as significant response of BPH patients towards 5ARIs were some of the findings that prompted large scale clinical trials to further investigate the possible role of these 5ARIs in the prevention or treatment of prostate cancer.
The prostate cancer prevention trial (PCPT) a multicenter, randomized, double blind, placebo-controlled clinical trial compared the finasteride with placebo in the prevention of adenocarcinoma of the prostate in men over the age of 55 and had a normal digital rectal examination. Thousands of men with a prostate-specific antigen level at or below 3.0 ng/mL, were randomly assigned to finasteride, (5 mg/day). The study showed decrease of 24.8% in prevalence of biopsy-proven prostate cancer finasteride treated population (18.4%) as compared with placebo (24.4%) over a period of 7 years. The PCPT findings were found to be very interesting with a less common urinary symptoms and significant improvement in term of increased flow rates and decreased prostate specific antigen level in the finasteride treated group, thus suggested possible role for 5ARIs in the chemoprevention of prostate cancer [7,34].
Further, because of the known influence of neoadjuvant hormone therapy in reducing the incidence of HGPIN [35]. It was further suggested that finasteride, an oral agent with no impact on serum testosterone levels, may be a useful agent for treating HGPIN. Thus in PCPT trials, men were evaluated for HGPIN, if they had a diagnosis of HGPIN or invasive cancer on an interim or end of study biopsy, or if they had a completely negative biopsy after seven years studies [36]. Men were diagnosed and examined jointly with HGPIN alone or HGPIN concurrently with invasive prostate cancer and observation indicated that men receiving finasteride had a significant 21 percent lower risk of HGPIN (9.2 versus 11.7 percent, hazard ratio 0.79, p-0.001). Based upon a previous studies of histological evaluation, where blockade of 5-alpha reductase with finasteride has not been found to cause obvious change in the morphology of prostate cancer but unlike other forms of androgen deprivation therapy, the effects of finasteride on the morphology of HGPIN and Gleason score are unknown [37].
Though finasteride effectively reduces the development of prostate cancer over a seven-year period in PCPT trials, however, adenocarcinomas with a high-grade appearance were found to be more common in the finasteride group (37 versus 22 percent). The public health impact of PCPT, and particularly whether finasteride promotes high-grade prostate cancer or not remains is a subject of intense ongoing debates [6].
Number of investigators hypothesized that a dual inhibition of both isozymes would lead to greater reduction of both plasma and prostatic DHT, therefore, greater clinical efficacy [23]. Variation of C-17 amide substituent on the optimal 4-aza-3-oxo-androstane skeleton was of particular interest in the search of potent dual azasteroid inhibitor and resulted into the development of first dual 5ARI (Dutasteride).
Dutasteride: Dutasteride also belongs to class of 4-aza-steroids and chemical name is 17β- N- {2, 5- bis (trifluoromethyl) phenyl)}-3- oxo- 4-aza- 5α- androst- 1- ene- 17- carboxamide. It was approved by U.S. FDA in 2002, for the symptomatic treatment of BPH after significant serum DHT suppression (94.7%±3.3% ) in Phase II, double-blind, placebo controlled, comparative dose-ranging trial, greater than with finasteride (70.8±18.3%, p<0.001) [38]. Unlike finasteride, dutasteride has been reported to be a non selective competitive inhibitor of both 5AR type 1 and type 2 isozymes. At clinical dose of 0.5 mg/day it has decreases DHT levels >90% by forming a stable complex with a slow rate of dissociation constant [39].
Another important outcomes of the study by Gleave and colleagues, showed that men treated with dutasteride (doses 0.5 mg daily or higher) for 4 months prior to radical prostatectomy had significantly lower intraprostatic DHT concentrations, but had higher testosterone concentrations than in untreated prostatectomies [40]. Dutasteride was found to improve urinary flow rate, decrease the risk of acute urinary retention and need for surgery by reducing the size of enlarged prostate. Improved efficacy of dutasteride (0.5 mg/day) over finasteride (5 mg/day) in terms of symptom score, maximal urinary flow rate and quality of life had also been published by Kumar, et al. [32].
Observations by Iczkowski and colleagues [41] with significant decrease (mean 15% v. 24%, p=0.025), in cancer tissues from men who received pre-surgical treatment with dutasteride, compared with that from men treated with finasteride, sparked the interest of researchers to further explore its potential in reducing the risk of prostate cancer and its treatment by conducting another large stage clinical trials REDUCE [42].
The reduction by dutasteride of prostate cancer events (REDUCE) trial is another multicenter, randomized, international, placebocontrolled, double blind clinical trial initiated in 2003 to evaluate the ability of dutasteride to decrease the risk of biopsy detectable prostate cancer in men with moderately elevated PSA levels. In the REDUCE trial, 6,729 men were randomly assigned to dutasteride or placebo for 4 years. Men in the dutasteride group experienced 22.8% less incidence of biopsy-proven prostate cancer dutasteride group, as compared with placebo group, but no significant difference was found in the frequency of high-grade disease [4].
Further, in randomized phase III trial, more than two hundred men with isolated HGPIN on biopsy were assessed for PCa prevention, using dutasteride 0.5 mg versus active surveillance. With the primary end cancer-free survival (CFS), per-protocol prostate biopsies were performed at 6, 12, 24, and 36 months till completion of the study period of 3-years. The 3-year PCa-free survival was observed to be 43.6% in the surveillance and 49.6% in that of dutasteride group and conclusion was drawn that it did not lower the PCa detection rate, but did not worsen detected PCa characteristics in men with HGPIN [43].
Animal models, particularly mouse models, has been found to play a central role in the study of the etiology, prevention and treatment of human prostate cancer (PCa). While tissue culture models are also extremely useful in understanding the biology of PCa, but they cannot recapitulate the complex cellular interactions within the tumor microenvironment that play a key role in cancer initiation and progression. Over the past twenty years, number of investigations has been carried by different research groups to establish the role of 5ARI in the management of prostate cancer [44].
In animal models, dutasteride, but not finasteride, inhibited growth of Dunning R-3327H rat prostate tumors [45]. Whereas, in nude mice bearing LNCaP human prostate cancer xenografts, both finasteride and dutasteride has reduced tumor growth, although dutasteride was found to be more effective at an equimolar dose [46]. Further, in rats, finasteride significantly decreased androgensensitive tissue weights, but did not decrease Dunning R-3327H tumor growth [47]. In all these animal studies, finasteride and dutasteride administration began once tumor had been established; finasteride administration initiated before tumor implantation may be more efficacious. On the other hand, regardless of when finasteride treatment is initiated, prostate cancer cells may compensate for 5AR type 1 inhibition by increasing 5AR type 2 expressions and/or activity; thus, the dual inhibitory effect of dutasteride may offer an advantage over finasteride.
Tolerability and risks of 5α-reductase inhibitors
Decades of experience has taught us to resist the temptation of blowing trumpets, at early impressive results, considering them as breakthroughs and the jubilations was short lived by the observations of large scale clinical trials of PCPT and REDUCE.
The potential beneficial role of 5ARIs like finasteride in a primary preventive setting as shown by the PCPT is partly nullified by the increased incidence of sexual adverse effects compared with placebo. The most commonly reported side effects on finasteride long term usage are decreased libido, ejaculatory dysfunction or impotence, while some of the patients showed rashes and breast enlargement. However, there was no documented evidence of increased survival in the finasteride group. Despite that they reported a 27% increased risk of high grade in the finasteride group over a 7-year period (280 of 4,368 men; 6.4%) compared with the placebo group (237 of 4,692 men; 5.1%). Recently published report in the issue of “The Oncologist”, by Kao and colleagues [48] based on their observation in 1489 patients with cancer and BPH in the Taiwanese National Health Insurance registry, also showed that finasteride use is associated with an actual increase in risk of prostate cancer.
This difference in the rate of high-grade disease was seen within the first year of the study and grading bias i.e. histologic changes that mimic those of high-grade disease caused by androgen-deprivation therapy could be one of the possible explanations for this difference [49]. It was suggested that finasteride may induces high-grade tumors by reducing the level of intracellular dihydrotestosterone within the prostate. Evidences also supported that the prostate tumors that develop in men with low testosterone levels were worse outcomes, than the prostate cancers that develop in men with normal testosterone levels. Thus it was suggested, that finasteride selects for high-grade tumors by selectively inhibiting low-grade tumors [50]. However, because of these differences of opinion as to whether this effect occurs with finasteride, FDA in 2011 announced to revise the 5ARI class of drugs with their new safety information. Long term follow-up in these men and further laboratory research will be required to determine the reason for the association between finasteride and high grade prostate cancer.
Despite the dual inhibition of 5AR type 1 and 2, and subsequent significant lower serum and prostatic levels of DHT, the tolerability of dutasteride are similar to that of finasteride. Two year study with dutasteride revealed similar rates of impotence, decreased libido, ejaculation disorder, and gynecomastia as that of finasteride [5]. The role of dutasteride is more acceptable in the secondary preventive setting. Fleshner, et al. [51] reported a randomized double-blind, placebo-controlled multi-centric trial with hundred of patients with low volume localized prostate cancers, followed by an active surveillance, where dutasteride and placebo were administered randomly on 1:1 basis for three years. Prostate cancer progression of 38% and 48% in the dutasteride and control group was observed and concluded that dutasteride can be useful adjunct to active surveillance in the management of low-risk Prostate cancer. However, in contrast to the above observation, RP Murtola, et al. [52] in a retrospective review state that 5ARI usage of 4 years or more is associated with an increased incidence of high grade prostate cancers with worse progression. Also the use of dutasteride was associated with no reduction in risk of prostate cancer and an increased risk of renal cancer. These disappointing results are in line with several other randomized trials investigating the use of these drugs for chemoprevention.
Recent 2011 instructions by the FDA promoted and encouraged Monga, et al. [53] to conduct the, meta analysis of dutasteride alone or in combination with tamsulosin, to quantify its effect on detection of prostate cancer and increased incidence of high-grade prostate cancer. Glaxo Smith Kline sponsored meta analysis phase III randomized clinical trials, with a study duration of ≥2 years demonstrated that in a population with symptomatic BPH and/or at increased risk of prostate cancer, a statistically significant lower number of detectable prostate cancers was found in men taking dutasteride compared to control groups. But there were several limitations that need to be considered when interpreting these results [54].
Conclusion
Increasing incidences of prostate cancer demanded an effective strategy to check its unabated occurrences. Chemoprevention with 5AR (Finasteride and Dutasteride) seemed to be holding such potential by inhibiting conversion of T to DHT. Controversies still surrounds the probability of these agents in impairing prostate cancer. While monotherapy with 5ARIs has a minimal impact on prostate cancer, there is much interest with combining the well tolerated hormonal effects of 5AR inhibitors with other therapeutic modalities. Additional studies are warranted to weigh the possible benefits and a reduced risk of urinary problems against sexual side effects and the increased risk of high-grade prostate cancer, and also to determine a safe, efficacious, cost-effective chemo preventive agent to be given to young men, for preventing the disease in the ageing population. Well designed randomized clinical trials addressing questions in these areas will hopefully lead to more efficacious and appropriate treatment of prostate cancer with lower disease specific mortality, while minimizing treatment related morbidity.
References
- Atlanta GA. American Cancer Society. Cancer Facts & Figures 2016: American Cancer Society, 2016.
- Ries L, Harkins D, Krapcho M, et al. National Cancer Institute.
- American Cancer Society. Cancer Facts and Figures.
- Hudak SJ, Hernandez J, Thompson IM. Role of 5 alpha-reductase inhibitors in the management of prostate cancer. Clin Interv Aging. 2006; 1: 425-431.
- Joseph CK, Ranko M, Cristina MG, Eric AK. High-Grade Prostatic Intraepithelial Neoplasia Korean. J Urol. 2012; 53: 297-303.
- Debra L Z, Ximing Y. High-grade Prostatic Intraepithelial Neoplasia of the Prostate: The Precursor Lesion of Prostate Cancer. Int J Clin Exp Pathol. 2009; 2: 327-338.
- Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003; 349: 215-224.
- Ahmed S, Lindsey B, Davies J. Emerging minimally invasive techniques for treating localized prostate cancer. BJU Int. 2005; 96: 1230-1234.
- Carter HB, Walsh PC, Landis P, Epstein JI. Expectant management of nonpalpable prostate cancer with curative intent: preliminary results. J Urol. 2002; 167: 1231-1234.
- Bartsch G, Rittmaster RS, Klocker H. Dihydrotestosterone and the concept of 5alpha-reductase inhibition in human benign prostatic hyperplasia. Eur Urol. 2000; 37: 367-380.
- Kenny B, Ballard S, Blagg J, Fox D. Pharmacological options in the treatment of benign prostatic hyperplasia. J Med Chem. 1997; 40: 1293-1315.
- Partin AW, Rodriguez R. The molecular biology, endocrinology, and physiology of the prostate and seminal vesicles. In: Walsh PC, Retik A, Vaughan E, et al. Campbell’s Urology. 8th ed. Philadelphia: Saunders; 2002. pp 1237-1296.
- Schweikert HU, Tunn UW. The metabolism of androstenedione and testosterone to C19 metabolites in normal breast, breast carcinoma and benign prostatic hypertrophy tissue. Steroids. 1987; 50: 19.
- Trachtenberg J, Hicks LL, Walsh PC. Androgen and estrogen receptor content in spontaneous and experimentally induced canine prostatic hyperplasia. J. Clin. Invest. 1980; 65: 1051-1059.
- Bruchovsky N, Wilson JD. The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. J Biol Chem. 1968; 243: 5953-5960.
- Blohm TR, Laughlin ME, Benson HD, Johnston JO, Wright CL, Schatzman GL, et al. Pharmacological induction of 5 alpha-reductase deficiency in the rat: separation of testosterone-mediated and 5 alpha-dihydrotestosteronemediated effects. Endocrinology. 1986; 119: 959-966.
- Jønler M, Riehmann M, Bruskewitz RC. Benign prostatic hyperplasia. Current pharmacological treatment. Drugs. 1994; 47: 66-81.
- Griesinger G, Felberbaum R, Diedrich K. GnRH-antagonists in reproductive medicine. Arch Gynecol Obstet. 2005; 273: 71-78.
- Ai N, De Lisle RK, Yu SJ, Welsh WJ. Computational model for predicting the binding affinity of ligands for the wild type androgen receptor and a mutated variant associated with human prostate cancer. Chem Res Toxicol 2003; 16: 1652-1660.
- Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem 2005; 48: 2972-2984.
- Bullock TL, Andriole GL Jr. Emerging drug therapies for benign prostatic hyperplasia. Expert Opin Emerg Drugs. 2006; 11: 111-123.
- Guess HA, Heyse JF, Gormley GJ. The effect of finasteride on prostatespecific antigen in men with benign prostatic hyperplasia. Prostate. 1993; 22: 31-37.
- Larry G, Alan So, Neil F, Ricardo R, Darrel D, Mostafa E. The role of 5-alpha reductase inhibitors in prostate pathophysiology: Is there an additional advantage to inhibition of type 1 isoenzyme? Can Urol Assoc J. 2009; 3: S109-S1014.
- Marks LS. 5a-Reductase: History and clinical importance. Rev Urol. 2004; S11-S21.
- Holt DA, Levy MA, Metcalf BW. Inhibition of Steroid 5R-Reductase. In Advances in Medicinal Chemistry. Maryanoff BE, Maryanoff CA. Eds; JAI Press Inc: Greenwich; 1993. 1-29.
- Abell AD. Henderson BR. Steroidal and Non-Steroidal Inhibitors of Steroid 5R-Reductase. Curr. Med. Chem. 1995; 2: 583-597.
- Occhiato EG, Guarna A, Danza G, Serio M. Selective non-steroidal inhibitors of 5 alpha-reductase type 1. J Steroid Biochem Mol Biol. 2004; 88: 1-16.
- Sun J, Xiang H, Yang LL, Chen JB. A review on steroidal 5α-reductase inhibitors for treatment of benign prostatic hyperplasia. Curr Med Chem. 2011; 18: 3576-3589.
- Andriole G, Bruchovsky N, Chung LW, Matsumoto AM, Rittmaster R, Roehrborn C, et al. Dihydrotestosterone and the prostate: the scientific rationale for 5alpha-reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004; 172: 1399-1403.
- Faragalla J, Bremner J, Brown D, Griffith R, Heaton A. Comparative pharmacophore development for inhibitors of human and rat 5-alphareductase. J Mol Graph Model. 2003; 22: 83-92.
- Tian G, Mook RA, Moss ML, Frye SV. Mechanism of time-dependent inhibition of 5 alpha-reductase by delta 1-4-azasteroids: Toward perfection of rates of time-dependent inhibition by using ligand-binding energies. Biochemistry 1995; 34: 13453-13459 .
- Kumar VL, Wahane VD. Current status of 5alpha-reductase inhibitors in the treatment of benign hyperplasia of prostate. Indian J Med Sci. 2008; 62: 167- 175.
- Rittmaster RS, Lemay A, Zwicker H, Capizzi TP, Winch S, Moore E, et al. Effect of finasteride, a 5 alpha-reductase inhibitor, on serum gonadotropins in normal men. J Clin Endocrinol Metab. 1992; 75: 484-488.
- Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003; 349: 215-224.
- Balaji KC, Rabbani F, Tsai H, Bastar A, Fair WR. Effect of neoadjuvant hormonal therapy on prostatic intraepithelial neoplasia and its prognostic significance. J Urol. 1999; 162: 753- 757.
- Yang XJ, Lecksell K, Short K, Gottesman J, Peterson L, Bannow J, et al. Does long-term finasteride therapy affect the histologic features of benign prostatic tissue and prostate cancer on needle biopsy? PLESS Study Group. Proscar Long-Term Efficacy and Safety Study. Urology. 1999; 53: 696-700.
- Bostwick DG and Qian J. Effect of androgen deprivation therapy on prostatic intraepithelial neoplasia. Urology. 2001; 58: 91-93.
- Marihart S, Harik M, Djavan B. Dutasteride: a review of current data on a novel dual inhibitor of 5alpha reductase. Rev Urol. 2005; 7: 203-210.
- Miller J, Tarter TH. Update on the use of dutasteride in the management of benign prostatic hypertrophy. Clin Interv Aging. 2007; 2: 99-104.
- Gleave M, Qian J, Andreou C , Pommerville P, Chin J, Casey R, et al. The effects of the dual 5alpha-reductase inhibitor dutasteride on localized prostate cancer—results from a 4-month pre-radical prostatectomy study. Prostate. 2006; 66: 1674-1685.
- Iczkowski KA, Qiu J, Qian J, Somerville MC, Rittmaster RS, Andriole GL, et al. The dual 5-alpha-reductase inhibitor dutasteride induces atrophic changes and decreases relative cancer volume in human prostate. Urology. 2005; 65: 76-82.
- Andriole GL, Humphrey P, Ray P, Gleave ME, Trachtenberg J, Thomas LN. Effect of the dual 5alpha-reductase inhibitor dutasteride on markers of tumor regression in prostate cancer. J Urol. 2004; 172: 915-919.
- Milonas D, Auskalnis S, Skulcius G, Gudinaviciene I, Jievaltas M, Joniau S. Dutasteride for the Prevention of Prostate Cancer in Men With High-Grade Prostatic Intraepithelial Neoplasia: Results of a Phase III Randomized Open- Label 3-Year Trial. World J Urol. 2016.
- Brawley OW. The potential for prostate cancer chemoprevention. Rev Urol. 2002; 4 Suppl 5: S11-7.
- Xu Y, Dalrymple SL, Becker RE, Denmeade SR, Isaacs JT. Pharmacologic basis for the enhanced efficacy of dutasteride against prostatic cancers. Clin Cancer Res. 2006; 12: 4072-4079.
- Opoku-Acheampong AB, Nelsen MK, Unis D, Lindshield BL. The effect of finasteride and dutasteride on the growth of WPE1-NA22 prostate cancer xenografts in nude mice. PLoS One. 2012; 7: e29068.
- Canene AK, Lindshiel DBL, Wang S, Jeffery EH, Clinton SK, Erdman JW, et al. Combinations of tomato and broccoli enhance antitumor activity in dunning R3327-H prostate adenocarcinomas. Cancer Res. 2007; 67: 836-843.
- Paller CJ, Smith TJ. Finasteride and prostate cancer: a commentary. Oncologist. 2012; 17: 888-890.
- Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003; 349: 215-224.
- Yang XJ, Lecksell K, Short K, Gottesman J, Peterson L, Bannow J, et al. Does long-term finasteride therapy affect the histologic features of benign prostatic tissue and prostate cancer on needle biopsy? PLESS Study Group. Proscar Long-Term Efficacy and Safety Study. Urology. 1999; 53: 696-700.
- Musquera M, Fleshner NE, Finelli A, Zlotta AR. The REDUCE trial: chemoprevention in prostate cancer using a dual 5alpha-reductase inhibitor, dutasteride. Expert Rev Anticancer Ther. 2008; 8: 1073-1079.
- Shrinivas RP. Use of dutasteride in carcinoma prostate. Uroscan. 2014; 30: 123-124.
- John RS, Brett D, Evans AJ. Therapy-associated effects in the prostate gland Histopa thology. 2012; 60: 153-165.
- Monga N, Sayani A, Rubinger DA, Wilson TH, Su Z. The effect of dutasteride on the detection of prostate cancer: A set of meta-analyses. Can Urol Assoc J. 2013; 7: E161-E167.