
Case Report
Austin J Cancer Clin Res. 2021; 8(1): 1087.
Late Sequelae after Neuroblastoma-Associated Paraneoplastic Anti-Hu Syndrome in a 4-Year-Old Boy
Till A1, Spiegler J1, Callsen F1, Ketzer J1, Langer T1, Tafazzoli K2, Wunsch L2, Winkler B3, Hero B4, Rostasy K5, Wandinger K6 and Lauten M1*
1Department of Pediatrics and Adolescent Medicine, University Hospital Schleswig-Holstein, Germany
2Department of Paediatric Surgery, University Hospital Schleswig-Holstein, Germany
3Department of Pediatric Haematology and Oncology, University Medical Centre Hamburg-Eppendorf, Germany
4Department of Pediatric Haematology and Oncology, University Children’s Hospital of Cologne, Germany
5Department of Pediatric Neurology, University of Witten/Herdecke, Germany
6University Hospital of Schleswig-Holstein, Institute of Clinical Chemistry, Germany
*Corresponding author: Melchior Lauten, Department of Pediatrics and Adolescent Medicine, University Hospital Schleswig-Holstein, Ratzeburger Allee 160, 23562 Lubeck, Germany
Received: February 13, 2021; Accepted: March 11, 2021; Published: March 18, 2021
Abstract
Anti-Hu syndrome is a rare autoantibody associated paraneoplastic disease of the central and peripheral nervous system resulting in a variety of neurological symptoms. In pediatric patients it is described in the context of (ganglio) neuroblastoma associated Opsoclonus-Myoclonus Syndrome (OMS) and other paraneoplastic syndromes. The timely diagnosis of paraneoplastic autoimmunity in childhood is hampered by its rarity as well as by the diversity of clinical symptoms that may occur. We report a 4-year-old boy with gastrointestinal disorder and neurological symptoms due to neuroblastomaassociated anti-Hu syndrome. The patient stabilized under a multimodal oncologic, immunosuppressive and antiepileptic treatment regimen. Treatment of neuroblastoma was individually modified and especially the specific anti- GD2 post-consolidation therapy was substituted by oral cyclophosphamide maintenance therapy in order not to aggravate autoimmune encephalitis. At the age of 8 years, the boy has been in ongoing complete oncologic remission for two years after end of relapse treatment. However, he suffers from neurologic symptoms like focal epilepsy and late sequelae of the oncologic disease.
This case shows that treatment of paraneoplastic anti-Hu syndrome is challenging, encephalitis may persist long after oncologic remission and may lead to developmental delay and a variety of physical sequelae.
Keywords: Paraneoplastic neurological syndromes; ANNA-1; Opsoclonusmyoclonus syndrome; Chronic gastrointestinal pseudo-obstruction; Rituximab; Glucocorticoids; (ganglio) Neuroblastoma; Anti-Hu syndrome; Late sequelae
Introduction
Anti-Hu syndrome is a rare paraneoplastic disease of the central and peripheral nervous system with detection of antineuronal nuclear autoantibodies against Hu antigens (anti-Hu Ab). Anti-Hu Abs, also termed ANNA-1 (Anti-Neuronal Nuclear Antibody-1), recognize different neuronal nuclear RNA-binding domains (Hu antigens) encoding for proteins regulating messenger RNA and neuronal differentiation of the central and peripheral nervous system [1,2]. Tumor cells outside the nervous system may also express Hu antigens [3] and trigger immune response with cross-reacting anti- Hu Abs and lymphocytes. This results in a variety of paraneoplastic neurological syndromes presenting with encephalomyelitis, sensory neuropathy, chronic gastrointestinal pseudoobstruction, cerebellar degeneration and limbic encephalitis [4]. Anti-Hu syndrome in adults is predominantly associated with small cell lung carcinoma [5] and neurological symptoms often precede tumor diagnosis. In pediatric patients anti-Hu Abs are described in the context of (ganglio) neuroblastoma derived Opsoclonus-Myoclonus Syndrome (OMS) [6] and other paraneoplastic syndromes [7,8].
We report a pediatric patient with paraneoplastic intestinal pseudoobstruction and encephalitis due to anti-Hu syndrome associated with neuroblastoma.
Case Presentation
A four-year-old boy was presented with an 18 months history of failure to thrive, abdominal pain, diarrhea and vomiting after foodintake. In addition, gait ataxia, loss of upright walk and posture, paresthesia of lower extremities, dizziness and adynamia as well as intermittent opsoclonus characterized by conjugated choatic eye movements which had been observed by his parents for the last year, supporting the diagnosis of Opsoclonus-Myoclonus- Syndrome (OMS). Diagnostic work-up revealed a paralytic ileus with distended duodenum due to a right-sided prevertebral mass at the gastric output, measuring 30mm. Metaiodobenzylguanidine (MIBG) scintigraphy was negative, Fluorodeoxyglucose Positron Emission Tomography Computed Tomography (FDG-PET-CT) showed only very low uptake of FDG, serum Neurone Specific Enolase (NSE) was 22.3μg/l and urine catecholamine metabolites were negative. Histologically, a poorly differentiated neuroblastoma (according to the INPC, International Neuroblastoma Pathology Committee) [9], was confirmed (no amplification of MYCN, no deletion of chromosome 1p, no bone marrow involvement). Clinically, feeding via Percutaneous Enteral Gastrostomy (PEG) and jejunostomy (PEJ) was commenced.
Oncologic treatment was initiated according to the GPOHNB2004 protocol, intermediate risk. During the first weeks of treatment, the patient developed epileptic seizures progressing to status epilepticus during episodes of septic fever. Cranial Magnetic Resonance Imaging (MRI) was normal, but laboratory workup pointed at anti-Hu associated encephalitis (serum anti-Hu Ab titer 1:3,200; Cerebrospinal Fluid (CSF): intrathecal immunoglobulin synthesis, no pleocytosis). Four weeks later, anti-Hu Ab were detected in the CSF as well (titre 1:10). Electroencephalography (EEG) showed absence of sleep graphoelements and right-sided frontotemporal focal slowing. Anticonvulsive treatment with levetiracetam and oxcarbazepine was initiated. Immunosuppressive treatment was started with intravenous dexamethasone pulses (20mg/m²/day on days 1-3). After one N6- and two N5-cycles, the tumor was resected and chemotherapy was terminated due to post-hoc down staging to initial stage 2 (International Neuroblastoma Staging System, INSS, [10]). Treatment of the anti-Hu syndrome was continued with intravenous dexamethasone pulse therapies at three- to fourweekly intervals. In addition, Prednisolone (PDN) was administered intrathecally (10mg/dose) and Rituximab was given four times intravenously (375mg/m²) and once intrathecally (2mg/dose). Gait ataxia and paresthesia improved and PEG/PEJ tube could be removed due to sufficient oral food intake. Serum anti-Hu Ab titres decreased to 1:100 and CSF titer dropped from 1:10 to 1:2 within eight months, however, never became negative (Figure 1). Interestingly, intrathecal Rituximab seemed to have a pronounced effect.
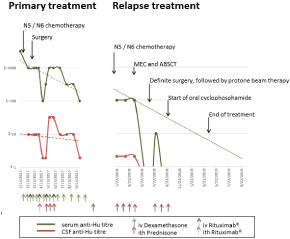
Figure 1: Anti-Hu titres and oncologic (upper part) as well as
immunosuppressive (lower part) systemic and intrathecal treatment. Partial
regression lines are given for anti-HU titres.
One year after initial diagnosis of neuroblastoma, walking difficulties, adynamia and dysarthria reoccurred and led to the diagnosis of neuroblastoma relapse at the interaortocaval level. Bone marrow showed positivity of GD2 (glycolipid disialoganglioside 2) positive cells (0.01%), leading to INSS stage 4 disease. Partial resection of the poorly differentiated neuroblastoma was performed and salvage therapy was initiated according the GPOH-NB2004 protocol, high risk arm [11], since response to N5/N6-cylces had been observed in first-line treatment. After three N5- and three N6-cycles, high dose chemotherapy Melphalan Etoposide Carboplatin (MEC) with autologous stem cell rescue (ABSCT) was performed, followed by complete surgical resection of the tumor and extracorporale protone beam irradiation of the former tumor region (36Gy in 20 fractions of 1.8Gy). Due to ongoing anti-Hu syndrome, we did not start post-consolidation state-of-the-art intravenous immunotherapy targeting GD2, but decided for oral maintenance chemotherapy with six N7 cycles (oral cyclophosphamide) [12]. In the further course, a dose reduction to two-third of cyclophosphamide became necessary due to bone marrow toxicity.
Intrathecal PDN treatment for the anti-Hu syndrome was continued during oncologic treatment and terminated as soon as oral cyclophosphamide was started.
The patient’s parents documented the intensity of residual paraneoplastic symptoms using a scale from 0 (normal/no symptoms) to 10 (severe symptoms). Symptom categories were mood, aggressiveness, speech/articulation, head posture, gait, abdominal complaints and bowel movements (Figure 1). Serum and CSF anti-Hu antibody titres during first-line treatment did not correlate with neurologic symptoms, however, they did during relapse treatment (Figure 2). Neurological and gastrointestinal symptoms improved with a combination of oncologic, immunosuppressive and antiepileptic medication (Figure 1) and serum anti-Hu antibody titres were negative after ABSCT and final tumor resection (Figure 2). Interestingly, dysarthria and ataxia improved within 2-3 weeks after every intrathecal treatment, however, especially intrathecal PDN was accompanied by untypically aggressive behaviour lasting for 1-2 weeks.
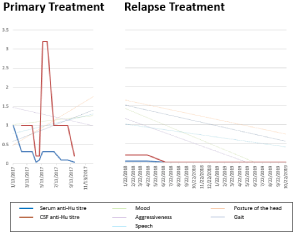
Figure 2: Anti-Hu titres and clinical symptoms during treatment. Titres and
clinical symptom scores are shown relative to the initial value at first evaluation.
The first value given was arbitrarily set to 1 (100%) and all following values
are calculated in relation to their corresponding initial values. A symptom
score of 0 corresponds to best mood, no aggressiveness, normal speech,
normal posture of the head and normal gait, as estimated by the patient’s
parents. In order not to overload the figure, we chose partial regression lines
to show the development of clinical symptoms.
Almost two years after completion of relapse neuroblastoma therapy, the eight-year-old boy remains in complete oncologic remission. He receives levetiracetam and lacosamide for focal epilepsy with break-through seizures during febrile infections. Additional neurological symptoms with changing intensity are ataxia, reduced peripheral reflexes and sensoneurinal hearing loss. Furthermore, he developed secondary Fanconi´s syndrome, chronic diarrhea due to pancreatic insufficiency and thoracic scoliosis. In addition, he shows infection related fatigue. Due to secondary immunodeficiency following Rituximab (low IgG levels, normal CD19+ cells) he receives monthly immunoglobulin substitution. The patient shows developmental delay and visits a specialized school with focus on motor development.
EEG currently shows persisting abnormal activity, predominantly in the right frontotemporal area. Two years and nine months after first neurologic symptoms of anti-Hu syndrome, cranial MRI revealed a both sided oblong enhancement of the external capsula without accumulation of contrast. This has been described in the context of anti-Hu syndrome.
Discussion/Conclusion
Gastrointestinal dysmotility in the context of anti-Hu syndrome presumably occurs due to lymphocytic infiltration [13]. Pathogenetically, anti-Hu antibody-induced loss of ganglion cells within the myenteric plexus is discussed [8]. Clinical symptoms indicating paraneoplastic anti-Hu autoimmunity may vary and often precede the diagnosis of oncologic disease [8,14,15]. In our patient, gastrointestinal disorder was misinterpreted, neurological symptoms were not recognized and diagnosis of neuroblastoma was delayed.
The timely diagnosis of paraneoplastic autoimmunity in childhood is hampered by its rarity and by the diversity of clinical symptoms. Apart from the neurological and gastrointestinal symptoms seen in our patient, additional neurologic disorders like sensory neuropathy, limbic or brainstem encephalitis, cerebellar degeneration or myelitis may first hint at paraneoplastic anti-Hu syndrome [4]. Until today, prognosis of anti-Hu syndrome remains critical with respect to long term sequelae including physical and neuropsychological symptoms [16,17].
Treatment of the oncologic disease is of major importance for the treatment of paraneoplastic syndromes [5]. However, treatment of OMS may successfully be supported by intravenous glucocorticoids [18,19] and autoimmune encephalitis may benefit from intrathecal glucocorticoids [20]. In case of non-response, immunosuppression may be intensified by Cyclophosphamide [20] or Rituximab [16,21,22]. The capability of Rituximab to cross the blood brain barrier is poor, however, its safety after intrathecal administration is acceptable [23,24], especially if administered over a few minutes with a maximum dose of 10mg dissolved in sodium-chloride 0.9% [25]. Side effects, nevertheless, include seizures, papilledema, cauda equine syndrome and encephalitis [24], all of which were not observed in our patient.
Overall, our patient clinically stabilized under a multimodal treatment including oncologic, immunosuppressive and antiepileptic therapy. Intravenous application of dexamethasone clinically did not sufficiently suppress autoinflammation, leading to escalation of immunosuppressive treatment using systemic and intrathecal Rituximab plus intrathecal PDN.
Serum and CSF anti-Hu antibody titres remained positive after completion of first-line treatment for neuroblastoma. During relapse treatment, anti-Hu antibodies became negative within one month after ABSCT with MEC conditioning and definite surgery. Usually applied immunotherapy with anti-GD2 antibodies was substituted by oral Cyclophophamide aiming at continuing immunosuppressive therapy for autoimmune encephalitis while maintaining anti-tumour effect. Immune reconstitution after ABSCT may have helped to eradicate the autoreactive lymphocyte clones and led to immune recalibration in the lymphocyte repertoire as has been described for other autoimmune diseases, e.g. multiple sclerosis [26].
Almost two years after completion of second-line neuroblastoma treatment the patient remains in complete remission. Late sequelae include treatment refractory focal epilepsy, ataxia, scoliosis and persistent diarrhea which, however, benefits from substitution of pancreatic enzymes. In addition, the patient suffers from gross-motor problems, intermittent fatigue, and developmental delay necessitating attendance of a specialized school.
This case shows that treatment of paraneoplastic anti-Hu syndrome is challenging, antibody titres do not correlate with clinical symptoms, encephalitis may last long after oncologic remission and may lead to neuropsychological late sequelae. A standard medical treatment is urgently needed to improve neurological and neuropsychological outcome of these patients.
References
- Giannopoulou C. Navigating the paraneoplastic neurological syndromes. Eur J Nucl Med Mol Imaging. 2003; 30: 333-338.
- Musunuru K, Darnell RB. Paraneoplastic neurologic disease antigens: RNAbinding proteins and signaling proteins in neuronal degeneration. Annu Rev Neurosci. 2001; 24: 239-262.
- Dalmau J, Furneaux HM, Rosenblum MK, Graus F, Posner JB. Detection of the anti-Hu antibody in specific regions of the nervous system and tumor from patients with paraneoplastic encephalomyelitis/sensory neuronopathy. Neurology. 1991; 41: 1757-1764.
- Zuliani L, Graus F. Neuronal nuclear antibodies, type 1 (Hu). Editors. In: Shoenfeld Y, Gershwin ME, Meroni PL. Antibodies. second edition: Elsevier. 2007: 185-190.
- Graus F, Keime-Guibert F, Rene R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001; 124: 1138-1148.
- Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, et al. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. Journal of pediatric hematology/oncology. 2000; 22: 315-320.
- Fisher PG, Wechsler DS, Singer HS. Anti-Hu antibody in a neuroblastomaassociated paraneoplastic syndrome. Pediatric neurology. 1994; 10: 309- 312.
- Drukker CA, Heij HA, Wijnaendts LC, Verbeke JI, Kaspers GJ. Paraneoplastic gastro-intestinal anti-Hu syndrome in neuroblastoma. Pediatric blood & cancer. 2009; 52: 396-398.
- Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999; 86: 364-372.
- Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993; 11: 1466-1477.
- Berthold F, Faldum A, Ernst A, Boos J, Dilloo D, Eggert A, et al. Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol. 2020; 31: 422-429.
- Berthold F, Boos J, Burdach S, Erttmann R, Henze G, Hermann J, et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with highrisk neuroblastoma: a randomised controlled trial. Lancet Oncol. 2005; 6: 649-658.
- De Giorgio R, Bovara M, Barbara G, Canossa M, Sarnelli G, De Ponti F, et al. Anti-HuD-induced neuronal apoptosis underlying paraneoplastic gut dysmotility. Gastroenterology. 2003; 125: 70-79.
- Aagre SV, Patel A, Choudhary M, Kataria P, Baldaniya K. Paraneoplastic encephalitis as a first evidence of recurrent neuroblastoma: A rare case entity. J Pediatr Neurosci. 2015; 10: 404-407.
- Wildhaber B, Niggli F, Stallmach T, Willi U, Stauffer UG, Sacher P. Intestinal pseudoobstruction as a paraneoplastic syndrome in ganglioneuroblastoma. Eur J Pediatr Surg. 2002; 12: 429-431.
- Blaes F, Dharmalingam B. Childhood opsoclonus-myoclonus syndrome: diagnosis and treatment. Expert Rev Neurother. 2016; 16: 641-648.
- Hero B, Schleiermacher G. Update on pediatric opsoclonus myoclonus syndrome. Neuropediatrics. 2013; 44: 324-329.
- Ertle F, Behnisch W, Al Mulla NA, Bessisso M, Rating D, Mechtersheimer G, et al. Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatric blood & cancer. 2008; 50: 683-687.
- Rostasy K, Wilken B, Baumann M, Muller-Deile K, Bieber I, Gartner J, et al. High dose pulsatile dexamethasone therapy in children with opsoclonusmyoclonus syndrome. Neuropediatrics. 2006; 37: 291-295.
- Bien CG, Bien C. Therapeutisches Management von Autoimmun- Enzephalitiden. Z Epileptol. 2015; 28: 201-206.
- Shams’ili S, de Beukelaar J, Gratama JW, Hooijkaas H, van den Bent M, van’t Veer M, et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol. 2006; 253: 16-20.
- Morales La Madrid A, Rubin CM, Kohrman M, Pytel P, Cohn SL. Opsoclonusmyoclonus and anti-Hu positive limbic encephalitis in a patient with neuroblastoma. Pediatric blood & cancer. 2012; 58: 472-474.
- Schulz H, Pels H, Schmidt-Wolf I, Zeelen U, Germing U, Engert A. Intraventricular treatment of relapsed central nervous system lymphoma with the anti-CD20 antibody rituximab. Haematologica. 2004; 89: 753-754.
- Jaime-Perez JC, Rodriguez-Romo LN, Gonzalez-Llano O, Chapa-Rodriguez A, Gomez-Almaguer D. Effectiveness of intrathecal rituximab in patients with acute lymphoblastic leukaemia relapsed to the CNS and resistant to conventional therapy. Br J Haematol. 2009; 144: 794-795.
- Bromberg JE, Doorduijn JK, Baars JW, van Imhoff GW, Enting R, van den Bent MJ. Acute painful lumbosacral paresthesia after intrathecal rituximab. J Neurol. 2012; 259: 559-561.
- Massey JC, Sutton IJ, Ma DDF, Moore JJ. Regenerating Immunotolerance in Multiple Sclerosis with Autologous Hematopoietic Stem Cell Transplant. Front Immunol. 2018; 9: 410.