
Research Article
Austin J Cancer Clin Res. 2021; 8(1): 1090.
MYBL1 Knockdown in a Triple Negative Breast Cancer Line: Evidence of Down-Regulation of MYBL2, TCF19 and KIF18b Expression
Player A*, Abraham N, Abdulrahman N, Nsende E, Cunningham S and Rogers S
Department of Biology, Texas Southern University Houston TX, USA
*Corresponding author: Audrey Player, Department of Biology, Texas Southern University, Houston Texas, USA
Received: May 21, 2021; Accepted: June 12, 2021; Published: June 19, 2021
Abstract
Purpose: The MYBL1 gene is a strong transcriptional activator, associated with cell cycle signaling and differentiation. Data show the gene is overexpressed in triple negative breast cancers. Considering the possibility that MYBL1 might be involved in events associated with the pathogenesis of these cancers, we sought to identify genes associated with MYBL1 expression in triple negative breast cancer.
Methods: shRNA lentiviral knockdown was used to down-regulate the MYBL1 gene. Microarray analyses were used to identify genes either directly or indirectly affected by targeting MYBL1 knockdown. Data analyses was performed utilizing Affymetrix TAC 4.0, Chip X transcription factor analyses, Target Scan miRNA analyses, and STRING analyses was used to determine protein: protein interaction and pathway analyses. Web Gestalt and Gene Ontology were used to determine pathway and gene-set enrichments. Publicly available patient and cell line datasets were retrieved and processed using resources available in Gene Expression Omnibus and Oncomine. The polymerase chain reaction and western analyses were used to determine transcript and protein levels, respectively.
Results: Knockdown of MYBL1 in a triple negative breast cell line led to down-regulation of MYBL2, TCF19, KIF18b along with an enrichment of cell cycle signaling genes. Gene expression analyses show that MYBL1, MYBL2, TCF19 and KIF18b display a similar pattern of expression in breast cell lines and many of the archival patient datasets examined.
Conclusion: TNBC is a heterogeneous subtype, so these data suggest that cancers that over-express MYBL1, express MYBL2, TCF19 and KIF18b. Bioinformatic analyses suggest MYBL1 regulates MYBL2 which leads to regulation of TCF19 and KIF18b.
Keywords: Triple negative breast cancer; MYBL1 knockdown; Microarray
Abbreviations
CCNB1-cyclin B1; CEL-microarray cell intensity file; Chipchromatin immunoprecipitation; DMEM: Dulbecco’s Modified Eagle Minimum Essential Media; E2F-E2F: Transcription Factors; ERBB2-Erb-B2: Receptor Tyrosine Kinase 2; ER: Estrogen Receptor; GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase; GEO: Gene Expression Omnibus; GSE: Gene Set Expression; FOXM1: Foxhead Box M1; KIF18b: Kinesin Family Protein18b; LIN: Linc Complex; LV: Lentiviral Particles; miRNA: microRNA; MYBL: Myeloblastosis Viral Oncogene-Like Protein; PCR: Polymerase Chain Reaction; PGR: Progesterone Receptor; RBL: Retinoblastoma-Like Proteins; RBBP4: Retinoblastoma Binding Protein 4; shRNA: small hairpin RNA; STRING: Search Tool for the Retrieval of Interacting Genes/ Proteins; TCF19: Transcription Factor 19; TFDP: Transcription Factor Dp; TNBC: Triple Negative Breast Cancer
Introduction
Triple Negative Breast Cancer (TNBC) is characterized as negative for three molecular signature genes, Estrogen Receptor (ER), Progesterone Receptor (PGR) and ERRB2. Even though the cancer is classified as an individual subtype, it is incredibly heterogeneous. Analyses of TNBC show that the sub type can be further classified into seven sub-categories [1]. Mostly, the TNBC are aggressive, they grow quickly, have a high recurrence rate and there are a limited number of treatment options for patients compared to patients with receptor positive breast cancers. Patients presenting with receptor positive gene expression are treated with hormone or targeted gene therapies; because TNBC slack the positive receptors, they do not respond to these therapies [2]. TNBC patients basically rely upon chemotherapy and radiation therapies. For this reason, the cancers are studied with the aim of identifying genes that might eventually be considered as potential targets for therapy.
The goal of our studies is to make a contribution towards characterizing TNBC. In an effort to characterize TNBC we performed comparative analyses of microarray datasets generated using cell lines and patient samples. Results from an earlier study [3] showed a differential pattern of expression of the MYBL1 gene in a subpopulation of TNBC compared to some luminal and most non-tumor breast samples. MYBL1 belongs to the MYB family of genes which includes c-MYB and MYBL2. The genes are protooncogenes that function as strong transcriptional activators involved in proliferation, differentiation and cell cycle signaling processes, all of which are associated with tumor progression [4-7]. The MYB proteins share substantial homology in their DNA binding domains [8] which allow for regulation of some of the same gene targets. The genes also contain distinctly different regions which allow for variations in post-translational modifications, which can ultimately lead to recognition and subsequent activation of different gene targets and biological activities [9]. Unique to MYBL1, the gene is a master regulator in the meiosis phase of the cell cycle in testis, demonstrating high levels in normal testes undergoing spermatogenesis [10]. Our studies have focused on characterizing MYBL1 in TNBC in an effort to ultimately determine the role of the gene in the pathogenesis of the cancers.
In addition to our studies, others show MYBL1 over-expression in luminal breast cancers [9] and the rare triple negative Breast Adenoid cystic carcinomas [11]. In one particular study, Liu et al [12] examined breast cancers via microarray and performed Supervised Network Analyses with the goal of determining the prognostic significance of over-expression of c-MYB and MYBL1 in receptor positive cancers. The investigators identified MYBL1 and nine other genes associated with poor prognosis in the receptor positive samples. In addition to breast cancers, MYBL1 is identified as over-expressed in colon cancers [13], uterine leiomyomas [14], murine B-cell lymphomas [15] and Burkitt’s lymphoma cells [16]. Considered together, these data support the study of MYBL1 for a possible role in cancers. Data show that changes in MYBL1 expression is caused by amplification, rearrangement and translocation events, with translocations involving NFIB and yet unspecified genes and mechanisms [11].
Based on our earlier observations and those by other investigators, we chose to study the MYBL1 gene in TNBC, with a focus on defining genes affected by its expression. Our immediate approach was to knockdown the MYBL1 gene in TNBC, followed by microarray and data analyses to identify genes coordinately dysregulated by the process. Preliminary results from this study are presented here. As expected, a significant portion of the candidate genes affected by decreasing MYBL1 expression were enriched in cell cycle signaling. Knockdown of MYBL1 led to a substantial decrease in MYBL2 expression as well. This observation is consistent with those by Rushton et al [9], which show that MYBL1 and MYBL2 are coexpressed and can activate some of the same genes more-so than any combination including the other family member, c-MYB.
A list of novel genes is identified as affected by the MYBL1 knockdown in the current study, the focus will describe the experimental validation of MYBL1, MYBL2 and two other genes, Transcription Factor 19 (TCF19) and Kinesin family protein 18b (KIF18B) genes.TCF19 and KIF18b are down-regulated following targeted knockdown of MYBL1 and down-regulation of MYBL2. Both TCF19and KIF18b have documented involvement with cell cycle signaling [17,18], in addition, data suggest the genes are involved in tumor progression which make them intriguing candidates to study in breast cancers as they relate to MYBL1. To our knowledge, this is the study to describe knockdown of MYBL1 in TNBC and the first documentation of a possible relationship between MYBL1, MYBL2, TCF19 and KIF18B. The relationship between the genes is unclear, although preliminary interpretations of the data suggest a close associationbetweenMYBL1 and MYBL2, and between MYBL2, TCF19 and KIF18b. Three miRNAs were up-regulated after MYBL1 knockdown and chosen as differentially expressed candidates as well. The miRNAs are selected based on differential expression and following Target Scan analyses [19] which predicts binding to either MYBL1 or MYBL2 transcripts. The small RNA results are presented in the current study, along with the experimental validation of the pattern of expression of MYBL1, MYBL2, TCF19 and KIF18b in a panel of breast cancer samples. The data show that similar to the results in the knockdown microarray, MYBL1, MYBL2, TCF19 and KIF18b show a co-ordinate pattern of expression in many of the breast cancer cell lines and patient samples.
Materials and Methods
Cell Lines, cell culture and patient sample datasets
MDA MB231, MCF7 and MCF10A cell lines were obtained from Atcc.org (Manassas, VA, USA) and used within one year of purchase. MDA MB231 represents an aggressive TNBC, MCF7 cells represent the luminal A subtype, and MCF10A cells represent a non-tumor triple negative (TN) cell type. The cells are cultured in Dulbecco’s Modified Eagle Minimum essential media (DMEM) (Millipore, Sigma, St. Louis, MO, USA) supplemented with 1% penicillin and 10 % serum (FBS) in a 37°C and 5% CO2 as suggested by the supplier. The cells were feed twice weekly and passaged when the cultures reached 90% confluence using a 0.25% trypsin solution (Millipore, Sigma, St. Louis, MO, USA). Patient and cell line datasets were retrieved from Gene Expression Omnibus (GEO; GSE65194, GDS2250, GSE12777, GSE29327)[20] and Oncomine.org [21]. The MCF10a controls were extracted from the GSE29327 dataset and combined with the GSE12777 dataset. The Affymetrix spiked controls in both datasets were compared and found to generate an almost exact correlation, allowing for the samples in the two datasets to be combined.
shRNA knockdown of MYBL1 in MDA MB231 cells
The MYBL1 shRNA Lentiviral particles and the scramble control particles were purchased from Origene (Cat # TL303089V; Rockville Md, USA). Four MYBL1 target specific particles (packaged from the pGFP-C-shLenti vector; labeled LVA, LVB, LVC, LVD) were supplied by Origene and screened for their efficiency to suppress expression of the MYBL1 transcript. The lentiviral particles were transduced into MDA MB231 TNBC cells (at a MOI of 10:1) and screened to determine the sequence most effective at down-regulating the MYBL1 transcript. MDA MB231 cells were incubated with the targeted or scrambled viral particles for 72 hours in the presence of polybrene (sc-134220; Santa Cruz Biotechnology, Dallas TX, USA) in the complete cell culture media. Lentiviral particles were removed and fresh media was added to the cells. The transduced cells were selected following growth in 1ug/ml puromycin (CAS 53792; Santa Cruz Biotechnology, Dallas TX, USA). LVA particles, identified by the TCTGATCCTGTAGCATGGAGTGACGTTAC sequence, demonstrated the most significant down-regulation of MYBL1 mRNA, as a result this preparation was used for the future experiments. Cells transduced with LVA and the scrambled control sequence were maintained in the presence of puromycin.
RNA extraction and cDNA generation
In order to screen for the knockdown of MYBL1, total RNA was extracted from cells previously transduced with scrambled control or the MYBL1 sequences, and compared to RNA extracted from untreated cells. Total RNA was isolated using the Trizol reagent (Life Technologies Inc., Grand Island, NY) as suggested by the manufacturer. The quantity and quality of the RNA was determined via spectrophotometer and 3-N-Morpholinopropanesulfonic acid (MOPS) gel electrophoresis, respectively.
The cDNA was generated using the iScriptTM Reverse Transcriptase kit supplied by BioRad (Hercules, CA, USA) according to the manufacturer’s suggestion. This material was used for the down-stream Polymerase Chain Reaction (PCR) to determine the relative difference in gene expression levels between the various samples.
Primer design and PCR analyses
The current laboratory has access to breast cancer cell line transcriptomes processed using the Affymetrix U133 plus 2.0 microarrays. Information from these datasets were used as an indication of the gene expression levels of genes that were not treated with the lentivirus. These data along with GEO archival data and data generated from the knockdown datasets were crosscompared and utilized as part of the screening process used to identify candidate genes for the current study. Nucleotide sequences for the genes were retrieved from the NetAffxTM resources (http:// www.affymetrix.com/estore/analysis/index.affx) available at Affymetrix.com (Thermo Fisher Scientific, Waltham Mass). The PCR primers were designed using the Primer 3TM software [22], using the default program conditions. The gene specific primer are GAPDH (forward) TCCCTGAGCTGAACGGGAAG and (reverse) GGAGGAGTGGGTGTCGCTGT; MYBL1 (Affymetrix probe-set 213906_at) (forward) TGGATAAGTCTGGGCTTATTGG and (reverse) CCATGCAAGTATGGCTGCTA; MYBL2 (Affymetrix probe-set (201710_at) (forward) GAGGGGGTCTGTGAATCTGA and (reverse) CCATCCTAAGCAGGGTCTGA;TCF19(Affymetrix probe-set (223274_at) (forward) TCTTAGGGGAAGGGGAGAGA and (reverse) GTCACAGCCATCACACTGGT; KIF18b (Affymetrix probe-set 222039_at) (forward) GCTCTTTTCCCCACCTGTCT and (reverse) TTGGAAATCAAGGCACCATT. The PCR reaction was performed using the AmpliTaqGoldTM master mix reagent following the instructions outlined by the manufacturer (Thermo Fisher, Waltham Mass, USA). The PCR products were analyzed on a 2% agarose gel, and image analyses were performed using the LiCor (Lincoln NE, USA) gel analysis system.
Microarray and Data analyses
Microarray hybridizations of the knockdown preparations were performed at the University of Texas Southwest Core Facility (UTSW; Dallas Texas, USA). Total RNA samples were shipped overnight to UTSW. aRNA was prepared and hybridized to the Affymetrix Clarion microarray gene-chip. The CEL intensity files were made available, and data analyses was performed in our laboratory using the Affymetrix TAC 4.0 software (Thermo Fisher Scientific, Waltham Mass). CEL intensity results were normalized using RMA and the differentially expressed genes were generated following Limma Bioconductor analyses. Coding transcripts that displayed at least a 4-folddifference in expression between the targeted compared to the scrambled sequence were selected for analyses. A lower differential expression threshold (of 2x) was used for selection of the miRNAs, as the miRNAs were also screened via Target Scan analyses [19] to identify their predicted nucleotide targets. The p-values were not generated by the TAC 4.0 program as two Clarion microarrays were hybridized and subsequently processed to identify the differentially expressed genes. Web Gelstalt [23] and Gene Ontology [24] analyses were used to determine gene-set enrichment, pathway and functional analyses of differentially expressed genes identified in cell lines and patient samples. Transcript plots and analyses were performed using Microsoft Excel. Molbiotools.com was used to compare the gene lists (http://www.molbiotools.com/listcompare.html).Transcription factor enrichment analyses was performed using the multi-omics Chip-X Enrichment Analyses 3.0 (ChEA3) [25] platform which combines Chip-Seq evidence from ENCODE, ReMap, publications, GTEx, ARCHS4 and Enrichr libraries. Transcription factor binding was also assessed using the Signaling Pathway Project [26]. Protein: Protein Interactive (PPI) Network analyses were performed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRINGTM; [27]).
Western Blotting
Whole cells were prepared in Ripalysis buffer (Santa Cruz Biotechnology, Dallas TX, USA) and the supernatants were processed, electrophoresed and probed for detection of specific proteins using protocols available at Novus Biologics (Novus Biologics Littleton CO). The primary antibody concentrations and incubation times were determined following the recommendation of the suppliers.
Antibodies: Actin was used at a 1:100 dilution (NB600-501SS; Novus Biologicals LLC, Littleton CO), and MYBL1 was used at a 1:500 dilution (sc-514682; Santa Cruz Biotechnology, Santa Cruz CA). MYBL2 was used at a 1:500 dilution (sc-81192; Santa Cruz Biotechnology, Santa Cruz CA), TCF19 was used at a 1:100 dilution (sc-390923; Santa Cruz Biotechnology, Santa Cruz CA), andKIF18b was used at a dilution of 1:1000 (A303-982A; Bethyl Laboratories, Montogomery TX, USA).Secondary HRP conjugated Anti mouse (HAF007; R and D Systems, Minneapolis, MN) and Anti Rabbit (NBP-2-30348H; Novus Biologicals LLC, Littleton CO) antibodies were used at a dilution of 1:4000. Western blotting results were visualized with the Clarity Western ECL substrate (Bio-Rad, Hercules, CA, USA) on a LICOR digital imaging system (LI-COR Biotechnology, Lincoln, NE).
Results
Screening and validation of MYBL1 shRNA lentiviral preparations
The aim of this current study is twofold. The first aim is to further characterize TNBC and because of an interest in MYBL1, the second is to identify genes directly or indirectly associated with MYBL1 expression in TNBC. In order to accomplish these aims, our approach was to knockdown the MYBL1 gene and examine the genes affected by this process. Four shRNA lentiviral sequences targeted for MYBL1, corresponding to different regions of the transcript, were purchased from Origene.com, and screened to determine the sequence that effectively decreased MYBL1 transcript levels. The LVA sequence was the most effective at down-regulating the MYBL1 gene (Figure 1), as a result, this preparation was used for all of the future experiments. Before committing to the microarray analyses, the LVA MYBL1 and scrambled control sequences were examined further for their effect in MDA MB231 cells. The LVA particles were effective at knockdown of both the MYBL1 transcript (Figure 2a) which ultimately led to downregulation of the protein (Figure 2b) in MDA MB231 cells compared to the scrambled and untreated sample preparations.
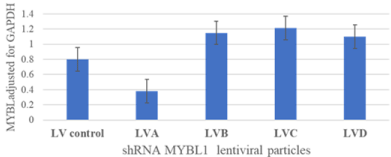
Figure 1: shRNAL entiviral particles screened for the ability to knockdown
expression of MYBL1 transcript. Various shRNA sequences were screened
for the ability to knockdown the MYBL1 sequence. The LVA shRNA
demonstrated adequate knockdown. The scramble non-targeting sequence
served as an off target negative control.

Figure 2: The LVA shRNA knockdownsMYBL1 transcript and subsequent
down-regulation of protein in the MDA MB231 cell preparations. The
transcripts were processed via PCR and the proteins via western analyses.
Cells transduced with the scrambled sequence and the untreated cell
preparations served as negative controls.
Results obtained following analyses of the microarray
Bioinformatic analyses of the microarrays following MYBL1 knockdown: Total RNA preparations isolated from the scrambled and knockdown preparations were examined via microarray, and the data was processed using the TAC 4.0 software. A total of 19 genes and 3 small RNAs were selected as candidate genes following knockdown of MYBL1. The candidate genes were identified based on (a) the degrees of differential gene expression (b) comparative analyses to expression in untreated microarray cell lines and patient samples in both our laboratory and GEO and (c) various bioinformatic analyses. Network Clustering analyses of the gene list show the genes are enriched in “Cyclin D associated G1/Polo-kinase mediated events” with an enrichment (i.e., observed/expected) ratio of 1.7 and a false discovery rate of 0.0025. The Gene Ontology Molecular Function analyses show the genes enriched in RNA polymerase II recruitment activity with an enrichment factor of 2.47 and a false discovery rate of 0.0045 [27]. Because of the size of the final gene list, reliable KEGG or Reactome pathway results were not generated by the various programs, although the results show clear evidence of association with the Cell cycle signaling pathway. The differentially expressed candidate genes represent transcripts and small RNAs both down and up-regulated following knockdown of MYBL1.The MYBL1, MYBL2, TCF19 and KIF18b transcripts demonstrate substantial down-regulation (Figure 3a) following targeted MYBL1 knock down. c-MYB, ER, PGR and ERRB2 are included as negative controls as these transcripts are negative in MDA MB231 cells and are not influenced by MYBL1 knockdown. Aside from MYBL1, MYBL2, TCF19 and KIF18b, other genes down-regulated following MYBL1 knockdown are plotted in Figures 3b-3c. Genes in Figure 3d were up-regulated following MYBL1 knockdown. Genes designated with an asterisk (*) have potential MYB family regulatory binding sequences as determined by the Signaling Pathway Project [26] and GeneCard.org analyses [28].
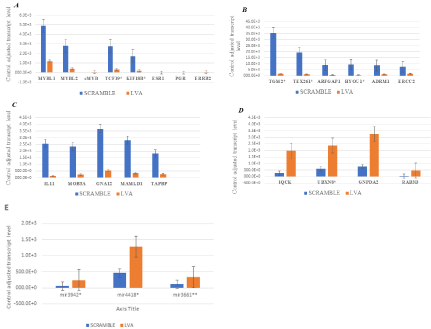
Figure 3: Control adjusted transcript levels of candidate genes differentially expressed following knockdown of MYBL1 in TNBC cells. (a-c) Genes that are downregulated
following knockdown of MYBL1 are demonstrated. (d) Genes that are up-regulated following knockdown of MYBL1. (e) The miRNAs plotted here were
up-regulated following knockdown of MYBL1. miRNAs designated with an asterisks (*) have predicted MYB gene regulatory sequences.
Small RNA analyses-Several small RNAs were also identified as affected by the MYBL1 knockdown. The miRNA3942, miRNA4418 and miRNA3661were up-regulated following knockdown of MYBL1 in MDA MB231 cells (Figure 3e). Target Scan prediction analyses predict binding of the miRNAs to either MYBL1 (*) or MYBL2 (**) transcripts.
Chip-X enrichment analyses and STRING analyses of candidate genes
Chip-X: The final candidate list includes19 genes. To determine their transcription factor enrichment, the candidate list was examined using the Chip-X online tool. Results of the analyses are listed in Table 1. MYBL1 ranks 11th out of 1632 transcription factor entries included in the Chip-X database; placing the gene in the top 0.7% of the entries. The rankings are generated by integrating information across the various libraries and are related to “how relevant the gene list is to the transcription factor” as determined by the Chip-X program. A lower ranking does not discount the involvement of the MYB transcription factor in regulating the candidate genes. So, these data support MYBL1 regulation of the genes on the candidate gene list. FOXM1 ranked first on the list, however the gene was not identified as differentially expressed in the current study. TP53 also ranked on the transcription factor enrichment list; the gene was differentially expressed but it was not selected for the current analyses. The candidate genes that ‘over-lap’ following the analyses between the various libraries are also designated in the table. Because of their citations across the different libraries, the data suggest, with some degree of confidence, that TCF19, KIF18b, MYBL2 and MYBL1 are coordinately regulated by the different transcription factors. A Cluster gram is generated directly from the data presented in Table 1. The Cluster gram analyses (Figure 4) show that of the candidate genes, MYBL2, TCF19, KIF18b and MYBL1 are the top candidates represented the most in the Chip-X libraries. These data also show that the four genes were cited across many of the same libraries.
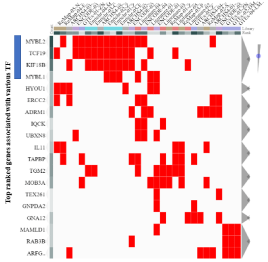
Figure 4: The Cluster gram was generated directly from data generated in
Table 1. Based on the list of differentially expressed genes, the results show
the top most citated genes across the various libraries.
![]()
Rank
Transcription factor
Score
Library
Overlapping genes
1
FOXM1
6.14E-04
ARCHS4 Coexpression
TCF19,KIF18B,MYBL2,HYOU1
2
ZNF107
6.22E-04
GTEx Coexpression
TCF19,MYBL2,MOB3A,TAPBP
3
ZNF107
7.12E-04
Enrichr Queries
TCF19,KIF18B,MYBL2,MYBL1
4
TP53
0.001229
ARCHS4 Coexpression
TCF19,GABARAPL1,ERCC2,MYBL2
5
ZNF331
0.001245
GTEx Coexpression
RAB3B,GABARAPL1,MAMLD1
6
ZNF367
0.001425
Enrichr Queries
TCF19,KIF18B,MYBL2,MYBL1
7
NPAS1
0.001843
ARCHS4 Coexpression
ADRM1,ERCC2,MYBL2
8
ZHX3
0.001867
GTEx Coexpression
GNPDA2,GNA12,TAPBP
9
CENPT
0.002137
Enrichr Queries
TCF19,KIF18B,MYBL2,TGM2
10
E2F7
0.002457
ARCHS4 Coexpression
TCF19,KIF18B,MYBL2
11
MYBL1
0.002849
Enrichr Queries
TCF19,KIF18B,MYBL2,TGM2
Table 1: Transcription factor enrichment analyses of the candidate gene list following Chip-X analyses. The differentially expressed genes were analyzed to determine transcription factor enrichment. These data predict an enrichment in FOXM1 regulation along with other transcription factors; MYBL1 is ranked #11 out of 1632 transcription factors in the database. TF=transcription factor. The libraries were generated from Chip-Seq datasets.
The MYBL2 transcription factor ranked #53 in the Chip-X assessment of our list of differentially expressed genes (data not shown), placing the transcription factor in the top 3.2% of the 1632 transcription factor entries. Even though MYBL2 ranks significantly lower than MYBL1, the results suggest the candidate gene list can be regulated by MYBL2. Based on these and other analyses, all four genes are considered together for further downstream experimental analyses in cell lines and breast cancer patient samples.
String
String is a functional protein interaction network. The protein interactive network analyses were performed in an effort to further examine a relationship between MYBL1, MYBL2, TCF19 and KIF18b gene candidates, and to identify additional genes associated with this particular network. This was accomplished by entering MYBL1, MYBL2, TCF19 and KIF18b into the STRING online search engine. The data show that MYBL1, MYBL2, TCF19 and KIF18b are closely related to CCNB1, LIN9, LIN37, KIF2C and FOXM1 via text mining, many related with a degree of confidence (Figure 5). The genes are all associated with cell cycle signaling, with strengths (i.e., observed/ expected ratios) ranging from 2.2-2.7 and false discovery rates ranging from 1.1 x 109 to 1012. Of the five new genes, only LIN37 was identified as differentially expressed gene on our microarrays. Again, FOXM1 is noted as associated with the MYBL1, MYBL2, TCF19 and KIF18b via a protein interaction network. The highest confidence interaction was between FOXM1 and MYBL2, and a slightly lower, but significant confidence interaction between FOXM1 and KIF18b gene.
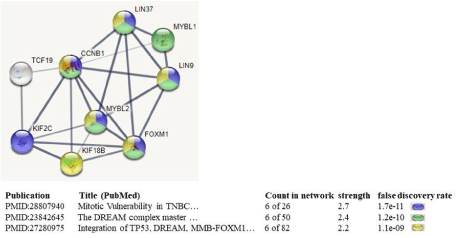
Figure 5: STRING analyses of the MYBL1, MYBL2, TCF19 and KIF18b: protein interaction network related to published data. The four genes were entered into
STRING to demonstrate additional genes more closely related to the four. KIF2C, FOXM1, CCNB1, LIN37 and LIN9 are cell cycle related genes, validating the
close relationship of our candidate genes with cell cycle related events. The thickness of the lines is ranked based on the highest confidence interactions (thick), to
high confidence interaction (less thick), to less confidence interactions (thin lines).
Experimental analyses of MYBL1, MYBL2, TCF19 and KIF18btranscript and protein levels in untreated breast cell lines: MYBL1, MYBL2, TCF19 and KIF18b gene expression levels are examined in cell lines and patient samples in an effort to determine whether the genes are coordinately expressed as observed following the bioinformatic analyses. For this portion of the study, the transcript and protein gene expression levels were experimentally determined in the untreated MCF10a, MCF7 and MDA MB231cell line preparations. The most dramatic differences in expression are between the MYBL1, MYBL2, TCF19 and KIF18btranscripts (Figure 6a) and proteins (Figure 6b) in TNBC compared to the MCF10a nontumor TN cell line. A similar pattern of expression for MYBL1 was observed in the previous study [3]. There appeared to be a difference between MYBL1in MCF7 compared to MDA MB231 cells. The two samples represent luminal A compared to TNBC, but because the heterogeneity of the subtypes, these data represent a preliminary, cursory assessment of the differences between the subtypes in this study. Consistent with the goal of this study, these data support data generated in the knockdown studies in that gene expression levels of MYBL1, MYBL2, TCF19 and KIF18b show a co-ordinate pattern of expression. It seems that both TCF19 and KIF18b are closely aligned with MYBL2 expression.
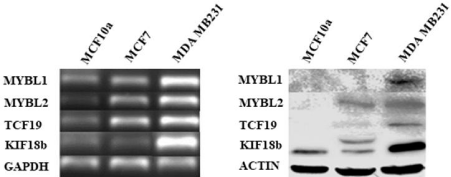
Figure 6: Analyses of transcript and protein levels in MYBL1, MYBL2, TCF19 and KIF18b in select cell lines.
(a)The MYBL1, MYBL2, TCF19 and KIF18b transcript levels were analyzed in MCF10a, MCF7 and MDA MB231 cell lines. (b)The MYBL1, MYBL2, TCF19 and
KIF18b protein levels were examined in MCF10a, MCF7 and MDA MB231 cell lines.
Analyses of MYBL1, MYBL2, TCF19 and KIF18b in GEO and Oncomine breast patient and cell line datasets: MYBL1, MYBL2, TCF19 and KIF18b transcript levels were examined in archived datasets generated from breast cancer patients and cell lines. The GEOGDS2250 dataset includes normal, luminal and basal-like/ TNBC patient samples (Figure 7a). Consistent with the experimental analyses, the greatest degree of differential expression was observed between non-tumor compared to the TNBC and, non-tumor compared to luminal breast cancer samples. A slight difference in expression was observed in luminal compared to TNBC patient samples, however, the difference was not statistically significant for this particular dataset. The GSE65194 data set included normal versus TNBC. These data show a significant difference in MYBL1, MYBL2, TCF19 and KIF18b expression in normal compared to TNBC patient samples (Figure 7b). Combined, the GSE12777 and GSE29327 datasets contained non-tumor, luminal and TNBC cell line samples which were analyzed for comparison of MYBL1, MYBL2, TCF19 and KIF18b gene expression. A similar pattern of expression was observed, in that the most significant pattern of differential expression for all four genes was observed in non-tumor compared to either the TNBC or the luminal breast cancer samples (Figure 7c).
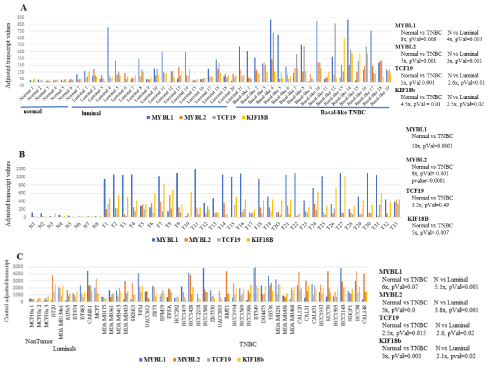
Figure 7: MYBL1, MYBL2, TCF19 and KIF18b analyses in GEO patient samples and cell lines.
(a) MYBL1, MYBL2, TCF19 and KIF18b transcript levels were determined in GDS2250 normal, luminal and basal-like/TNBC patient samples. (b)MYBL1, MYBL2,
TCF19 and KIF18b transcript levels were determined in GSE65194 normal and TNBC patient samples. (c)MYBL1, MYBL2, TCF19 and KIF18b transcript levels
were determined in GSE12777 non-tumor, luminal and TNBC cell lines. Significance values for differential expression are given. N=normal.
MYBL1, MYBL2, TCF19 and KIF18b transcript levels were also examined using the Oncomine breast datasets. Four datasets were retrieved and examined for expression of the four genes in normal versus breast cancer patients (Table 2). Affymetrix probesets corresponding to all four genes were present in the Richardson, TCGA and the Bittner datasets. The Richardson and TCGA samples showed increased expression in MYBL1, MYBL2, TCF19 and KIF18b genes in the cancers compared to the normal patient samples. In the Bittner dataset, MYBL2 was significantly differentially expressed in normal compared to cancer and MYBL1 was marginally overexpressed; TCF19 and KIF18b were not significantly differentially expressed in these patient samples. In the Yu dataset, only MYBL1 was differentially expressed; the dataset did not contain the TCF19 gene as determined by search of the probe-set ID. This microarray contains ~12K genes, and is a different analyses platform compared to the microarrays utilized in the Richardson, TCGA and Bittner studies. This could account for the observed differences. None of the analyses were stratified by molecular signatures, nonetheless, results of the analyses show the most consistent pattern of differential expression of the MYBL1 gene, followed by MYBL2, TCF19 and KIF18b gene expression. In conclusion, overall the results from the current analyses do not demonstrate perfect concordance between the expression of MYBL1, MYBL2, TCF19 and KIF18b but the degree of association between the expression of the genes warrants their continued analyses.
![]()
# Sample
Gene
p value
Fold difference
Ductal Carcinoma vs Normal (Richardson Breast 2, Cancer Cell 2006)
47
MYBL1
4x10-5
5x
MYBL2
2.7x10-7
5x
TCF19
1.7x10-9
4.1x
KIF18b
1.5x10-5
3.6x
Invasive Breast Carcinoma vs Normal, not published, 2011 TCGA
593
MYBL1
3.4x10-19
3.1x
MYBL2
8.7x10-9
3.8x
TCF19
1.1x10-12
2.0x
KIF18b
1.5x10-5
3.6x
Breast Adenocarinoma vs Adjacent Normal (Bittner Breast) 2004
257
MYBL1
1.9x10-7
1.96x
MYBL2
7.6x10-10
2.8x
TCF19
1.1x10-7
1.6x
KIF18b
1.5x10-6
1.6x
Breast Cancer vs Adjacent Normal, Yu Multi Cancer, Plos Genet, 2009
183
MYBL1
9X10-15
2.5X
MYBL2
0.328
1.0X
KIF18B
0.028
1.2X
Table 2: Oncomine dataset analyses of normal compared to breast cancer patients. The datasets were utilized to compare MYBL1, MYBL2, TCF19 and KIF18B gene expression analyses in normal vs breast cancer patients.
Discussion
To our knowledge this is the first study to knockdown MYBL1 in a TNBC cell line, determine genes affected by this process and examine expression of these genes in non-tumor TN, luminal and TNBC samples. Knockdown studies of the MYBL1 transcript can only address certain questions related to the possible relationship between the gene and TNBC, but these are questions that we see as essential towards beginning to understand how the gene works in breast cancer. Following the knockdown of MYBL1, a list of candidate genes was identified, including MYBL2. The differentially expressed genes were enriched in cell cycle signaling and in genes involved in RNA polymerase II transcription initiation accessory events. This is not remarkable, considering MYBL1 and MYBL2 are often co-expressed and have documented involvement in cell cycle processes. MYBL1 and MYBL2 were expected as candidates, however TCF19 and KIF18b were elected following gene expression analyses, and Chip-X and other bioinformatic analyses. Preliminary analyses suggest these genes are suitable candidates for further study in TNBC.
The ultimate genotype of cancers represents a hodgepodge of genes coordinately dysregulated that drive the tumors. We and others are considering the possibility that MYBL1 plays a role in this process. If so, then genes affected by MYBL1 also play a role, hence the inclusion of MYBL2, TCF19 and KIF18b to the mix, contributing their unique biological properties. In addition to MYBL1 and MYBL2, TCF19 functions as a transcription factor and regulates expression of events involved in tumor progression and KIF18b is a motor protein associated with events uniquely related to movement. We are not suggesting that these are the only players in this process, but because they are dysregulated, they should be considered for their contributions.
Knockdown of MYBL1 led to down-regulation of MYBL2 gene in this study. Both genes belong to the MYB family and are involved in cell proliferation and cell cycling processes [5,29-31]. Ziebold et al [32] performed one of the earliest experiments linking MYBL1 to the cell cycle process. Their study showed that MYBL1 is involved in G1 to S phase progression and activation is via phosphorylation of the C-terminus by cyclin dependent kinases. Related to their co-operativity, MYBL1 and MYBL2 are key proteins responsible for regulating the periodic expression of cell cycle signaling genes associated in the DREAM regulatory complex. The DREAM complex consists of interactions between MYBL1 and MYBL2, andE2F4, E2F5, LIN9, LIN37, LIN52, LIN54, RBL1, RBL2, RBBP4, TFDP1 and TFDP2 proteins [33,34]. In addition toMYBL1 and MYBL2, the E2F4, LIN37 and TFDP2 genes were also dysregulated in the current knockdown study (data not shown). With or without inclusion of E2F4, LIN37 and TFDP2 genes, analyses of the differentially expressed genes identified in our studies showed enrichment in genes associated with cell cycle signaling.MYBL1 and MYBL2 proteins have almost identical N-terminal DNA binding domains, hence they are able to bind some of the same targets and simultaneously regulate each other [29,35]. Rushton et al [7,9] show that MYBL1 and MYBL2 are co-expressed in breast samples, and can target and regulate the same genes. In addition to the gene expression data, protein interaction predictions suggest direct binding of MYBL1 to MYBL2 (Integrated Interactive Database [36].
The transcription factor enrichment analyses addressed whether the candidate genes showed enrichments in transcriptional regulation. Chip-X analyses suggest both enrichment of the gene list regulation by MYBL1 and MYBL2 transcription factors. In addition, results of these data show the most substantial enrichment by FOXM1 transcription factor regulation.FOXM1 is involved in cell proliferation and has been shown to regulate cyclin B1 and cyclin D1 and the timed expression of specific cell cycle phases [37-39]. Similar to MYBL1 and MYBL2, data show FOXM1 is associated with the regulatory network of the DREAM complex. FOXM1 is also overexpressed in breast cancers [40], and KEGG pathway analyses validate that both FOXM1 and MYBL2are key regulations of cellular senescence [41]. However, FOXM1was not affected by MYBL1 knockdown, but it could be that FOXM1 exerts its effects on genes indirectly related to MYBL1 or via regulation of MYBL2, TCF19 and/or KIF18b. The role and possible relationship between FOXM1 and MYBL1 in TNBC can be addressed by further analyses of our datasets and/or knockdown of FOXM1 in TNBC.
The TCF19 gene is involved in cell cycle progression, apoptosis and tumor progression [17,42]. Structurally, TCF19 gene has a N-terminal Fox Head Associated (FHA) domain and C-terminal Plant homeodomain (PHD) zinc finger domain. The PHD finger domain is shown to interact with methylated Histone H3 (H3K4me3) which suggests regulation via epigenetic processes. Similar to MYBL1, TCF19 is required for progression from G1 to S phase of the cell cycle [43]. Zeng et al [44] show the TCF19 gene is involved in cell proliferation and tumorigenesis in hepatocellular carcinoma, and Mondal et al [45] show TCF19 is necessary for both proliferation and survival of pancreatic tumor cells. siRNA-mediated knockdown of Tcf19 in the INS-1 insulinoma cell line, led to a reduction in numerous G1 to M phase cell cycle genes [17] further validating the involvement of TCF19 in cell cycle signaling. These data show TCF19 involvement in both cell cycle signaling and tumorigenesis.
TCF19 was chosen as part of this current study for several reasons. First, TCF19 is down-regulated by knockdown of MYBL1 in the TNBC cell line. Second, the gene demonstrates a similar pattern of expression with MYBL1 (and MYBL2) when compared to TNBC clinical samples and cell lines following bioinformatic analyses and experimental analyses of gene expression. The Cluster gram analyses of Chip-X show a reliable association with MYBL2 and to a lesser extent, MYBL1. Third, along with the key genes MYBL1 and MYBL2, TCF19isinvolved in cell cycle signaling. Lastly, the Signaling Pathway Project analyses (SPP) [26] demonstrate the MYB family of transcription factor scan regulate the TCF19 gene [46]. The SPP data do not confirm MYB regulation of TCF19; similar to all bioinformatic analyses, it only represents a prediction, which in this case is based on Chip-Seq binding assessment. We will continue to study the differentially expressed candidate genes, including TCF19 and KIF18b and their expression in efforts to further characterize the TNBC.
The KIF18b gene belongs to a superfamily of Kinesin genes that function in a vast number of cellular processes related to mitosis, meiosis and intracellular transportation [47]. The KIF18bgene is a motor protein which allows for chromosome movement during the mitotic phase of the cell cycle [18].An interaction between KIF18b and KIFl2cis required for microtubule depolarization [48]. KIF18b is related to tumor progression in cervical [49] and lung cancer [50] and contributes to the proliferation and metastasis of cutaneous melanoma [51]. More closely aligned with the current study, Li et al [52] performed a meta-analysis of the expression of the kinesin superfamily genes in breast cancer. The authors find that KIF18b is overexpressed in basal/TNBC and that over-expression of the gene correlates with worse overall survival; they recommend the gene be considered as a potential prognostic biomarker. The kinesin proteins are motor proteins unique in their ability to drive processes involving movement, which itself is a process related to tumor progression. The kinesin proteins are obvious candidates to study for their involvement in cancers. Data in this current study suggest that there is a relationship between KIF18band the MYB genes. Supporting our observations, previous data show that MYBL2 and FOXM1 bind to the promoter region and regulate the expression of several kinesin family genes [53]. It could be that targeed knockdown of MYBL1 in TNBC leads to down-regulation of MYBL2 which in turn leads to down-regulation of KIF18b.This suggestion is based on Chip-X analyses which show a closer relationship between MYBL2 and KIF18b compared to MYBL1. Like TCF19, aside from the genes’ known role in cell cycle signaling, it’s unclear as to the precise relationship between KIF18b and the MYB genes. The data suggest a closer relationship between MYBL2 and TCF19 and KIF18, however the results are somewhat confounded by the Chip-X transcription factor enrichment analyses which show a higher ranking and proposed association with MYBL1. The rankings represent statistically significant possibilities, but they are also related to the number of publicly generated citations. There is still a possibility that the TCF19 and KIF18b genes are directly regulated by MYBL1, but the association has to be determined experimentally.
A list of small RNAs were also affected by knockdown of MYBL1. The miRNAs include miR3942, miR4418 and miR3661. Each show at least a 2-fold increase in upregulation following knockdown of MYBL1. In addition, Target Scan prediction analyses suggest miR3942 and miR4418 are predicted targets for MYBL1 while miR3661 is a predicted target forMYBL2 transcript. There is essentially no biological data describing these miRNAs, except for miRNA3942 which is identified after sequencing normal and breast tumors [54].
Data presented here demonstrate preliminary analyses of the genes identified following the knockdown of MYBL1 in the MDA MB231 cell line. TNBC are heterogenous, so this study is not meant to define genes associated with MYBL1 in TNBCs in general. It is meant to be a preliminary analysis of the genes directly and/or indirectly associated with MYBL1 gene expression in TNBC cells. The current experiments will be expanded and further validated with a focus on understanding the relationship between MYBL1, MYBL2, TCF19 and KIF18b in TNBC.
Acknowledgment
The authors would like to thank Drs. Hector Miranda and Mario Hollomon for the use of their equipment during the course of this project, and the Department of Biology at Texas Southern University for their continued support.
References
- Lehmann BD, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121: 2750-2767.
- Bianchini G, et al. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016; 13: 674-690.
- Player A, et al. Identification of candidate genes associated with triple negative breast cancer. Genes Cancer. 2017; 8: 659-672.
- Marhamati DJ, et al. A-myb is expressed in bovine vascular smooth muscle cells during the late G1-to-S phase transition and cooperates with c-myc to mediate progression to S phase. Mol Cell Biol. 1997; 17: 2448-2457.
- Weston K. Myb proteins in life, death and differentiation. Curr Opin Genet Dev. 1998; 8: 76-81.
- Ma XP, Calabretta B. DNA binding and transactivation activity of A-myb, a c-myb-related gene. Cancer Res. 1994; 54: 6512-6516.
- Rushton JJ, Ness SA. The conserved DNA binding domain mediates similar regulatory interactions for A-Myb, B-Myb, and c-Myb transcription factors. Blood Cells Mol Dis. 2001; 27: 459-463.
- Nomura N, et al. Isolation of human cDNA clones of myb-related genes, A-myb and B-myb. Nucleic Acids Res. 1988; 16: 11075-11089.
- Rushton JJ, et al. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene. 2003; 22: 308-313.
- Bolcun-Filas E, et al. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development. 2011; 138: 3319-3330.
- Kim J, et al. MYBL1 rearrangements and MYB amplification in breast adenoid cystic carcinomas lacking the MYB-NFIB fusion gene. J Pathol. 2018; 244: 143-150.
- Liu LY, et al. A supervised network analysis on gene expression profiles of breast tumors predicts a 41-gene prognostic signature of the transcription factor MYB across molecular subtypes. Comput Math Methods Med. 2014; 2014: 813067.
- Guo H, et al. O-Linked N-Acetylglucosamine (O-GlcNAc) Expression Levels Epigenetically Regulate Colon Cancer Tumorigenesis by Affecting the Cancer Stem Cell Compartment via Modulating Expression of Transcriptional Factor MYBL1. J Biol Chem. 2017; 292: 4123-4137.
- Swartz CD, et al. Estrogen-induced changes in IGF-I, Myb family and MAP kinase pathway genes in human uterine leiomyoma and normal uterine smooth muscle cell lines. Mol Hum Reprod. 2005; 11: 441-450.
- Arsura M, et al. A-myb rescues murine B-cell lymphomas from IgM-receptormediated apoptosis through c-myc transcriptional regulation. Blood. 2000; 96: 1013-1020.
- Golay J, et al. Expression of A-myb, but not c-myb and B-myb, is restricted to Burkitt’s lymphoma, sIg+ B-acute lymphoblastic leukemia, and a subset of chronic lymphocytic leukemias. Blood. 1996; 87: 1900-1911.
- Krautkramer KA, et al. Tcf19 is a novel islet factor necessary for proliferation and survival in the INS-1 β-cell line. Am J Physiol Endocrinol Metab; 2013; 305: E600-E610.
- Lee YM, et al. Cell cycle-regulated expression and subcellular localization of a kinesin-8 member human KIF18B. Gene. 2010; 466: 16-25.
- Agarwal V, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4.
- Barrett T, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013; 41: D991-D995.
- Rhodes DR, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004; 6: 1-6.
- Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007; 23: 1289-1291.
- Liao Y, et al. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019; 47:. W199-w205.
- Mi H, et al. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019; 47: D419-d426.
- Keenan AB, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019; 47: W212-w224.
- Ochsner SA, et al. The Signaling Pathways Project, an integrated ‘omics knowledgebase for mammalian cellular signaling pathways. Sci Data, 2019; 6: 252.
- Szklarczyk D, et al. The STRING database in 2017: quality-controlled proteinprotein association networks, made broadly accessible. Nucleic Acids Res. 2017; 45: D362-d368.
- Zhang W, et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat Genet. 2018; 50: 613- 620.
- Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Front Biosci (Landmark Ed). 2011; 16: 1109-1131.
- Iness AN, et al. The cell cycle regulatory DREAM complex is disrupted by high expression of oncogenic B-Myb. Oncogene. 2019; 38: 1080-1092.
- Vaquerizas JM, et al. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009; 10: 252-263.
- Ziebold U, Klempnauer KH. Linking Myb to the cell cycle: cyclin-dependent phosphorylation and regulation of A-Myb activity. Oncogene. 1997; 15: 1011- 109.
- Rashid NN, Rothan HA, Yusoff MS. The association of mammalian DREAM complex and HPV16 E7 proteins. Am J Cancer Res. 2015; 5: 3525-3233.
- Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018; 25: 114-132.
- Ness SA. The Myb oncoprotein: regulating a regulator. Biochim Biophys Acta. 1996; 1288: F123-F139.
- Kotlyar M, et al. IID 2018 update: context-specific physical protein-protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019; 47: D581-d589.
- Chen X, et al. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol Cell Biol. 2013; 33: 227-236.
- Fischer M, Müller GA. Cell cycle transcription control: DREAM/MuvB and RBE2F complexes. Crit Rev Biochem Mol Biol. 2017; 52: 638-662.
- Fischer M, et al. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016; 44: 6070-6086.
- Koo CY, Muir KW, Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochim Biophys Acta. 2012; 1819: 28-37.
- Musa J, et al. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017; 8: e2895.
- Echevarría-Vargas IM, et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol Med. 2018; 10.
- Rolland T, et al. A proteome-scale map of the human interactome network. Cell. 2014; 159: 1212-1226.
- Zeng CX, et al. TCF19 enhances cell proliferation in hepatocellular carcinoma by activating the ATK/FOXO1 signaling pathway. Neoplasma. 2019; 66: 46- 53.
- Mondal P, et al. TCF19 Promotes Cell Proliferation through Binding to the Histone H3K4me3 Mark. Biochemistry. 2020; 59: 389-399.
- Mansour MR, et al. Analysis of the TAL1, MYB and MED1 cistromes in human MOLT3, Jurkat and HSPC cells, P. Signaling Pathways, Editor. 2014.
- Lucanus AJ, Yip GW. Kinesin superfamily: roles in breast cancer, patient prognosis and therapeutics. Oncogene. 2018.;37: 833-838.
- Oh CK, et al. Role of kif2c, A Gene Related to ALL Relapse, in Embryonic Hematopoiesis in Zebrafish. Int J Mol Sci. 2020; 21.
- Wu Y, et al. KIF18B promotes tumor progression through activating the Wnt/β-catenin pathway in cervical cancer. Onco Targets Ther. 2018; 11: 707- 1720.
- Ji Z, et al. KIF18B as a regulator in microtubule movement accelerates tumor progression and triggers poor outcome in lung adenocarcinoma. Tissue Cell. 2019; 61: 44-50.
- Yan H, Zhu C, Zhang L. Kinesin family member 18B: A contributor and facilitator in the proliferation and metastasis of cutaneous melanoma. J Biochem Mol Toxicol. 2019; 33: e22409.
- Li TF, et al. Overexpression of kinesin superfamily members as prognostic biomarkers of breast cancer. Cancer Cell Int. 2020; 20: 123.
- Wolter P, et al. Central spindle proteins and mitotic kinesins are direct transcriptional targets of MuvB, B-MYB and FOXM1 in breast cancer cell lines and are potential targets for therapy. Oncotarget. 2017; 8: 11160-11172.
- Persson H, et al. Identification of new microRNAs in paired normal and tumor breast tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res. 2011; 71: 78-86.