
Case Report
Austin Cardio & Cardiovasc Case Rep. 2019; 4(1): 1032.
Surgical Repair of Absent Pulmonary Valve with Double Outlet Right Ventricle in a 5 Year Old Child
Kar S*, Joshi R, Joshi RK, Agarwal M, Aggarwal N
Department of Pediatirc Cardiac Sciences, Sir Ganga Ram Hospital, New Delhi, India
*Corresponding author: Sibashankar Kar, Department of Pediatric Cardiac Sciences, Sir Ganga Ram Hospital, New Delhi, India
Received: April 15, 2019; Accepted: May 22, 2019; Published: May 29, 2019
Abstract
Absent Pulmonary valve with Double Outlet Right Ventricle is a rare association. Extensive literature search revealed only few reports. We report such a case along with situs inversus totalis and dextrocardia. The child was diagnosed at the age of 5 year and successfully underwent biventricular repair with routing of left ventricle to aorta by patch closure of ventricular septal defect and insertion of bileaflet pulmonary valve.
Keywords: Absent Pulmonary Valve; Double Outlet Right Ventricle; Absent Pulmonary Valve Syndrome
Introduction
Absent Pulmonary Valve (APV) syndrome is a rare congenital cardiac malformation consisting of dysplasia of pulmonary valve tissue, annular stenosis with severe incompetence of the valve, and dilatation of the Pulmonary Artery (PA) system, often leading to varying degrees of airway compression. It was first described by Chevers in 1847. In most patients, APV is sporadic with no identifiable genetic cause. This may be the result of error or complete failure of valve development.
APV is usually associated with Tetralogy of Fallot (TOF), several reports quoting 3-6% incidence, but association with Double Outlet Right Ventricle (DORV) is rare. The incidence of chromosome 22q11.2 deletion in APV with TOF ranges upto 75% but no study has been done linking it to DORV.
Case Presentation
A 5-year old male child, weighing 13.3Kg, presented with shortness of breath on exertion and cyanosis noted since late infancy. He was born at term, with adequate birth weight, to consanguineous parents. On examination, he demonstrated central cyanosis with SpO2 of 73% and grade-III clubbing. Cardiac examination revealed apex beat on the right fifth intercostal space lateral to the mid-clavicular line, a single S2, an ejection systolic murmur and early diastolic murmur. Routine blood investigations were normal except for polycythemia.
Chest X-ray (Figure 1) and ultrasound of abdomen showed situs inversus totalis. Echocardiography revealed an unrestrictive Ventricular Septal Defect (VSD) and aortic override of 70%, severe pulmonary stenosis with peak gradient of 80mmHg and severe pulmonary regurgitation (Figure 2A), dilatation of the main and branch PAs, dilated and hypertrophied Right Ventricle (RV) and aorto-mitral discontinuity due to a large subaortic conus measuring 9mm (Figure 2B). Cardiac angiography (Figure 2C) was done, as an institutional criteria, to confirm the echocardiographic finding of anomalous origin of Right Coronary Artery (RCA) from Left Coronary Artery (LCA). It also showed a retroaortic innominate vein. Branch pulmonary arteries Z-scores were + 5.3 and + 4.6 for right (18mm) and left (17mm) respectively.
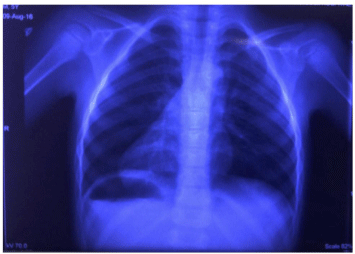
Figure 1: Chest X Ray showing abdominal situs inversus with dextrocardia.
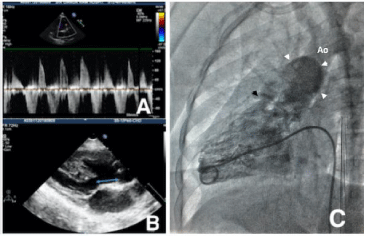
Figure 2: A) Doppler signal across right ventricular outflow tract suggestive
of pulmonary stenosis and pulmonary regurgitation, B) Subaortic conus
demarcated by double ended arrows, C) Right heart cath study showing the
narrow pulmonary annulus (black arrow head) and dilated pulmonary artery
(white arrow heads).
Patient was taken for surgical correction after informed consent from parents. After midline sternotomy, usual cardiopulmonary bypass was commenced. Left ventricle vented through left superior pulmonary vein. Approaching the patient from his left side, a right atriotomy was performed through which infundibular resection was done. Biventricular repair of DORV was achieved by intracardiac routing of VSD to aorta with a redundant Dacron patch (BARD Peripheral Vascular Inc., Tempe, Arizona, USA) using interrupted pledgeted 5-0 polypropylene. Care was taken to avoid the conduction system by placing the sutures farther than 5mm from the posteroinferior margin of the VSD.
Pulmonary arteriotomy was done, which revealed APV represented by scattered flat nubbins (Figure 3A) along the annular zone. Though the anomalous RCA was crossing the RVOT, a safe area was available between the pulmonary annulus and the RCA to extend the arteriotomy incision across the annulus into the distal infundibulum to accommodate a hegar appropriate opening. A 0.1mm non-porous polytetraflouroethylene membrane (BARD Peripheral Vascular Inc., Tempe, Arizona, USA) was sized and inserted to construct a bileaflet valve. This was overlaid with a liberal untreated autologous pericardial Transannular Patch (TAP) using 6-0 polypropylene (Figure 3B). Details of procedure have been illustrated in our published article [1].
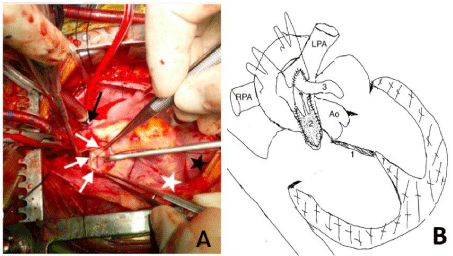
Figure 3: A) Stenotic and malformed pulmonary annulus with nonfunctioning
rudimentary nubbins(white arrows), aorta (black arrow), diaphragm (Black
star), dextrocardic right sided apex (white star). B) Schematic representation
of surgical procedure; VSD closure indicated by ‘1’, placement of bicuspid
valve by ‘2’ and transannular patch marked by ‘3’. Ao- Aorta, LPA- Left
Pulmonary Artery, RPA- Right Pulmonary Artery.
Post-operative course was uneventful and child was discharged in stable condition. Follow up at 30 month, child was asymptomatic and echocardiography showed normal functioning bileaflet pulmonary valves with no stenosis or regurgitation. Child was thriving well with normal growth and development. Oxygen saturation at room air was 98%.
Comments
Literature search for a combination of DORV with APV yield very few reports in scientific journals published in English language [2-8]. One case [2] was diagnosed in early infancy who died soon afterward and another case [3]
was diagnosed in the operation room who died 12 hours post operatively. Three cases [4-6] were diagnosed at prenatal necropsy. Successful surgical correction had been done in two reports [7,8]. Parikh et al [7] had unbalanced atrioventricular canal and had performed single ventricle palliation. Guo et al [8] had performed operation in a 14 year old female with additional bronchiarctia. Though bronchiarctia is not a surgical contraindication but will affect the period of recovery, hence we recommend an early surgical correction before development of respiratory symptom.
Our case was unique with simultaneous presence of situs inversus totalis and dextrocardia. Thymus was large and bilobed in our case making it unlikely for DiGeorge syndrome. Coronary anomaly was noted in our case in form of a single coronary arising from right posterior sinus and RCA arising from it and crossed the RVOT. Coronary anomaly was also reported in other case [3] who had rotation of coronary ostia to 90 degree from their normal location. Extracardiac anomalies were absent in our case but others [4,5] have noted in form of facial and limb deformities.
Cath study (Figure 2C) confirmed abnormal coronaries, narrow pulmonary annulus and dilated PAs. Our case did not have any significant airway obstruction that would otherwise have warranted bronchoscopy or CT scan. Intraoperatively we have noticed the aortic override of 70%, presence of subaortic conus and absence of aortomitral continuity, justifying the lesion being DORV rather than TOF.
Bileaflet PTFE pulmonary valve is sewn in such a way that it preserves growth potential of the pulmonary annulus. RV-PA valved conduit can be an alternative in coronary anomalies, but it does not have growth potential and often degenerates over time needing reintervention. Hence, we prefer handsewn bileaflet PTFE pulmonary valve as first option, whenever possible, to increase the quality of life and prevent the inadvertent shortcomings associated with RV-PA valved conduit. The overlaid TAP was comparatively narrow in our case because of the presence of already dilated MPA.
APV with DORV is a rare entity along with situs inversus totalis and dextrocardia. Anomalous coronary crossing RVOT further makes the surgery challenging. We believe, very little has been known on APV with DORV, which makes this association a promising area of research.
- Wankhade PR, Aggarwal N, Joshi RK, Agarwal M, Joshi R, Mehta A, et al. Short-term clinical and echocardiographic outcomes after use of polytetrafluoroethylene bicuspid pulmonary valve during the repair of tetralogy of Fallot. 2019; 12: 25.
- D’Cruz IA, Lendrum BL, Novak G. Congenital absence of the pulmonary valve. American Heart Journal. 1964; 68: 728-740.
- Baker WP, Kelminson LL, Turner WM, Jr., Blount SG, Jr. Absence of pulmonic valve associated with double-outlet right ventricle. Circulation. 1967; 36: 452- 455.
- Hartwig NG, Vermeij-Keers C, De Vries HE, Gittenberger-De Groot AC. Aplasia of semilunar valve leaflets: two case reports and developmental aspects. Pediatr Cardiol. 1991; 12: 114-117.
- Marek J, Skovranek J, Povysilova V. Congenital absence of aortic and pulmonary valve in a fetus with severe heart failure. Heart. 1996; 75: 98-100.
- Becker R, Schmitz L, Guschmann M, Wegner RD, Stiemer B, Entezami M. Prenatal diagnosis of familial absent pulmonary valve syndrome: case report and review of the literature. Ultrasound Obstet Gynecol. 2001; 17: 263-267.
- Parikh K, Muniz JC, Lopez L, Welch E, Drossner D, Sasaki NJC. Rare Occurrence of Absent Pulmonary Valve Syndrome in the Setting of Double- Outlet Right Ventricle with Unbalanced Complete Atrioventricular Canal Defect. 2019.
- Guo HC, Ren CW, Dai J, Lai YQ. Surgical Treatment of Double Outlet Right Ventricle with Absent Pulmonary Valve and Bronchiarctia. Chin Med J (Engl). 2017; 130: 881-882.