
Case Series
Austin Cardio & Cardiovasc Case Rep. 2022; 8(1): 1050.
A Case Series on Patients of Cardiac Amyloidosis with Varying Initial Clinical Presentation in a Tertiary Health Care Cardiology Outpatient Service, Kerala
Antony P Pathadan¹; Suresh Davis²; Ramdas Nayak¹; Jacob George¹; Sahala Abbas4*; Latha K Abraham³
1Department of Cardiology, Rajagiri Hospital, India
2Department of Cardiology, Diploma in Diabetology, Rajagiri Hospital, India
3Department of Pathology, Rajagiri Hospital, India
4Department of Cardiology, Medical Officer, Rajagiri Hospital, India
*Corresponding author: Sahala Abbas Department of Cardiology, Medical Officer, Rajagiri Hospital, Kerala, India.
Received: February 22, 2023 Accepted: March 29, 2023 Published: April 05, 2023
Abstract
The study aims to assess the outcome of early diagnosis in cardiac amyloidosis and to emphasize the need for high clinical index of suspicion even before onset of cardiac symptoms. Recent advances in immunohistochemical markers, molecular and genetic studies carry early diagnostic and grave prognostic significance. This study concludes a significant inverse relationship between time of diagnosis after the onset of cardiac symptoms and percentage survival rate of the affected individuals.
Introduction
Cardiac Amyloidosis (CA) has emerged as an underdiagnosed cause of Heart Failure (HF) that is associated with significant morbidity and mortality, particularly in later stages of disease [1,2]. Heart failure with amyloidosis is associated with higher odds of in-hospital mortality, 30-day readmission, and a longer length of hospital stay. It may be the presenting feature of the disease or may be identified while investigating a patient presenting with other organ involvement.
Small, single-centric studies have estimated the prevalence of Cardiac Amyloidosis as 13% among patients with {HF with preserved ejection fraction} [3,4]. Most cases of Cardiac Amyloidosis are of either transthyretin type, which may be acquired in older individuals or inherited in younger patients, or acquired monoclonal immunoglobulin light chain (AL) type.
Significant advances in non-invasive diagnostic testing [5,6] and targeted amyloid therapeutics [7] have piqued clinical enthusiasm; however, diagnostic delays of up to 34 months still persist [8,9]. Amyloidosis being a systemic disorder, biopsies can be obtained from several sites, including the heart {in ATTR -transthyretin amyloidosis}, due to predominant cardiac involvement), abdominal fat pad, bone marrow (as part of work-up for plasma cell dyscrasia in suspected AL amyloidosis), or kidney [1].
Prognosis in amyloidosis depends predominantly on the degree of cardiac involvement. Atrial fibrillation is common and is assosiated with poor prognosis. Though the prognosis is better in ATTR amyloidosis, both types of amyloidosis carry a high risk of mortality. As a result, timely diagnosis is critical to allow treatment initiation in earlier stages of disease, where inhibition of amyloid fibril formation has greater clinical benefit. Treatment for amyloidosis has evolved significantly over the past several years [7]. Treatment follows two parallel paths:
A) Treating the consequence of organ dysfunction and attempting to slow the progression of disease with chemotherapy against the plasma cells.
B) Cardiac specific treatment including {diuretics/salt restriction} and managing arrhythmias.
Currently available therapies include transthyretin stabilizers and transthyretin synthesis inhibitors for transthyretin amyloidosis, chemotherapy and autologous stem cell transplantation for light chain amyloidosis, and cardiac transplantation for selected patients with advanced HF [10]. ACE-Inhibitors, Angiotensin Receptor Blockers and Beta blockers are poorly tolerated and may result in profound hypotension. Pacemakers are frequently required due to assosiated conduction disease .As the disease is irreversible with high mortality, a cardiac transplant may be considered in indicated patients.
Methods
The study was a retrospective study with one year follows up of the patients conducted in the department of cardiology, Rajagiri hospital. The study subjects included all patients who were diagnosed with cardiac amyloidosis in Rajagiri hospital from time period August 2020 to August 2021. The relevant cinical information were obtained from electronic case report of the patient. Cinical details that were collected included age, gender, BMI(kg/m2), associated comorbidities, intial presenting symptoms and assessment of of liver, spleen and other organ involvement was done. The haemoglobin, serum creatinine, NT proBNP, 24 hour urine protein ratio, troponin, baseline LV function at the time of diagnosis was studied. Genetic study done by the patient was analasyed. Fat pad biopsy, immunohistochemistry slides of the patients available in department of pathology was studied for identifying the type of amyloidosis. The data was then entered in spread sheets of Microsoft Office Excel and the variables were compared quantitatively and qualitatively. The treatment details in relation to number of chemotherapy cycles [CyboR-D based chemotherapy), stem cell transplantation were collected from the department of hematology. The treatment follow of the patient was continued for a period of one year by simultaneously entering them in the spreadsheet and the results were tabularised as in (Table 1). These patients were furthur divided according to revised Mayo prognostic index 2012.
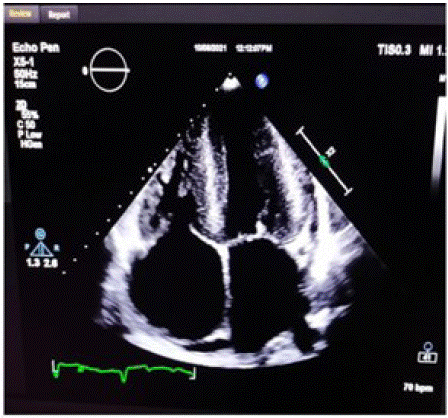
Figure 1: Echocardiogram of patient with cardiac amyloidosis showing biatrial enlargement.
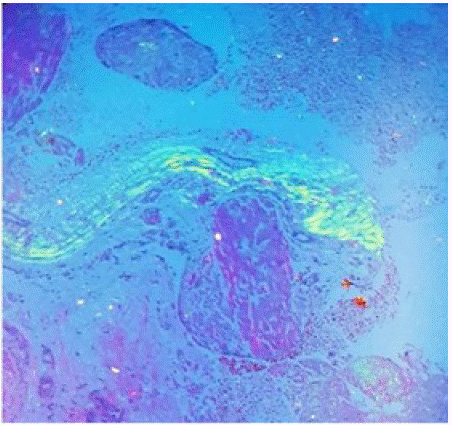
Figure 2: Apple green bifringence wit congo red stain of abdominal fat pad biopsy specimen.
![]()
Basic Characterstics
1
2
3
4
5
6
7
8
Age (years)
63 years
41 years
72 years
63 years
53 years
51 years
68 years
69 years
Gender
Female
Female
Male
Male
Female
Female
Male
Male
BMI (kg/m2)
26.3
26
16
63
21
22
22
26
Comorbidities
Nil
Nil
CAD ,
COPDCKD,
CLDDLP hypothyroidism
DM
Nil
Hypothyroidism Hypertension Dyslipidemia
Initial presentation
CCF
Arthralgia
weight loss
VT
CCF
weight
loss,
malaenaDifficulty
in walking
since
5years worsened
in past 1 year.Recurrent syncope,
limb
weakness
and
spasticity.Other
organ involvementliver
Shoulder joint , liver
Lung- type 2 respiratory failure
Gastroparesis-antral ulcers
liver ,spleen
Liver,
chronic proctitis, chronic deodinitisHypotonia Sensori-motor axonal polyneuropathy
gastric ulcerChronic
Sensori-motor polyneuropathy-Hemoglobin(g/dl)
10.2
11.8
13.4
10.4
13.2
11.4
15
12
Serum creatinine
0.8
0.9
1.2
2.7
4.4
0.9
0.7
1.2
NT proBNP
7096
553
5058
15,070
> 25,000
4311
279
500
Troponin
0.26
negative
0.12.
0.14
0.8
0.05
negative
negative
DFLC
(kappa -3.3-19) Lamda -5.7-26}Kappa-6
Lamda-448Kappa-6380
Lamda-9Kappa-20
Lamda-874Kappa-12
Lamda-4401Kappa -20
Lamda-133Kappa-1123
Lamda-35Kappa -17.44
Lamda -12.61Kappa -23
Lamda -12FreeKappa /lamda
ratio {0.26-1.65}0.01
708
0.02
0.002
0.15
31.4
1.38
1.9
- Hour Urine Protein (<150)
130
250
146
1785
5481
1689
NA
NA
Bone marrow plasma cells
(% nucleated marrow cells)39%
4%
2%
26%
4%
7%
2%
8%
Baseline LV function
60%
60%
55%
35%
40%
60%
60%
60%
Genetic study
Not done
Not done
Not done
Not done
Not done
Not done
TTR+ on exon 5 hetrozygous -heriditary amyloidosis {autosomal dominant}.
TTR+ on exon 5 hetrozygous -heriditary amyloidosis {autosomal dominant }.
PMP22(-) Exon5hetrozygous -heriditary amyloidosis {autosomal dominant}.Time from
onset
of symptoms
to
diagnosis (months)
2 months
4 months
1 year
2 year
4 months
8 months
1 year
8 months
Treatment regimen
(CyboR-D
based chemotherapy)Tolerated
12 cycles
of chemotherapyChemotherapy followed
by stem cell transplantationInitiated
on chemotherapy, but could not tolerateInitiated
on chemotherapy,
But couldn�t tolerateTolerated
12 cycles
of chemotherapyInitiated
on chemotherapyIntiated
on chemotherapyTolerated
12cycles
of chemotherapyOne year
follow up detailsOn maintainance
On mainatainance
Cardiac arrest
Cardiac arrest
On maintenance
On maintenance
On maintainance
On
mainatainance
Table 1: One year follow up details of the patients included in the study.
Discussion
A total of 8 patients were included in the study. The predominant age group affected was 60-70 years with mean age group as 60 years. Cardiac involvement in amyloidosis presents commonly with arrhythmias and/or rapidly progressing heart failure or occasionally with polyneuropathy, arthralgia or gastrointestinal symptoms. Once a patient develops cardiac symptoms, they usually have large amount of amyloid in the heart. Amyloid deposit in the atrium causes an irregular surface that makes these patients prone to thrombus formation, irrespective of atrial fibrillation development. Amyloid fibrils cause stiffening of heart which explains restrictive physiology found on echocardiogram.
Genetic study encouraged early diagnosis. Patients diagnosed with short duration of symptoms had good prognosis even though they presented in heart failure. Meanwhile patients diagnosed with long duration of symptoms had poor prognosis even though they were not in failure. A better treatment outcome in seen in patients who had stem cell transplantation following chemotherapy. The patients with assosiated multiorgan failure couldn’t tolerate chemotherapy and succumbed to death while the rest are on maintainance on one year follow up.
Thus we could see in our study, a significant inverse relationship between time of diagnosis after the onset of cardiac symptoms and their life expectancy rate and could conclude high clinical index of suspicion is needed for early diagnosis and good outcome.
Author Statements
Acknowledgement
The authors express sincere gratitude to Dr Mobin Paul, Department of Hemato Oncology, Rajagiri hospital for sincere guidance and moral support throughout the course of their work.
Funding
No funding sources
Conflict of Interest
None declared.
Informed Consent
Written informed consent was obtained from the patient.
References
- Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz, MA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016; 68: 1014-20.
- Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachman HJ, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018; 39: 2799-806.
- Gonzlez-L¢pez E, Gallego-Delgado M, Guzzo-Merello G, Haro-Del Moral FJ, Cobo-Marcos M, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015; 36: 2585-94.
- Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, et al. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. J Am CollCardiol HF. 2020; 8: 712-24.
- Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016; 133: 2404-12.
- Baggiano A, Boldrini M, Martinez-Naharro A, et al. Noncontrast magnetic resonance for the diagnosis of cardiac amyloidosis. J Am Coll Cardiol Img. 2020; 13: 69-80.
- Zhang KW, Stockerl-Goldstein KE, Lenihan DJ. Emerging therapeutics for the treatment of light chain and transthyretin amyloidosis. J Am CollCardiol Basic Trans Science. 2019; 4: 1-11.
- Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, et al. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018; 25: 174-79.
- Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light chain amyloidosis: patient experience survey from the Amyloidosis Research Consortium. Adv Ther. 2015; 32: 920-28.
- Barrett CD, Alexander KM, Zhao H, Haddad F, Cheng P, et al. Outcomes in patients with cardiac amyloidosis undergoing heart transplantation. J Am Coll Cardiol HF. 2020; 8: 461-68.