
Review Article
Austin J Cardiovasc Dis Atherosclerosis. 2018; 5(1): 1034.
Cardioprotection by Bioactive Polyphenols: A Strategic View
Chu AJ*
Department of Surgery, School of Medicine, Wayne State University, Detroit, USA
*Corresponding author: Arthur J. Chu, Department of Surgery, School of Medicine, Wayne State University, Detroit, USA
Received: February 15, 2018; Accepted: March 23, 2018; Published: March 30, 2018
Abstract
Cardiovascular disease (CVD) remains one causing most mortality worldwide. Common CVD risks include oxidative stress, hyperlipidemia, endothelial dysfunction, thrombosis, hypertension, hyperhomocysteinemia, inflammation, diabetes, obesity, physical inactivity, and genetic factors. Among which, lifestyle changes including diets are modifiable CVD risks, becoming the first line of prevention prior to any medication. In the past decades, the USDA, the American Heart Association, the American Nutrition Association, the Academy of Nutrition and Dietetics, and many other health organizations have launched five colors daily with vegetable and fruit consumption for human health. Lipophilic polyphenols, phytochemicals rich in vegetables and fruits, show classical antioxidation (e.g., radical-scavenging, metal chelating, NOX inhibition, attenuation on mitochondrial respiration, inhibition on xanthine oxidase, and upregulations on endogenous antioxidant enzymes), multiple effects on cell signaling (e.g., AMPK activation, SirT1 activation, eNOS activation, FOXO activation, NFkB inactivation, PI3K/AkT inhibition, mTORC1 inhibition, ERK inhibition, JAK/STAT inhibition, IKK/JNK inhibition, PDE inhibition, a-catenin inactivation, downregulation on TLR expression, ACE inhibition, adiponectin elevation, attenuated ET-1 production, and K+ channel activation), and many other biological actions (e.g., inhibition on a-glucosidase, anticoagulation, upregulation on paraoxonase 1, PAI-1 downregulation, tPA upregulation, epigenetic modulation, and altered gut microbiota). Accordingly, polyphenols multiple-targeting CVD risks and progression (Graphic summary) could offer broad range of cardioprotection from atherosclerosis, hypertrophy, arrhythmia, angina, heart failure, etc.
Keywords: Polyphenol; Anti-Oxidation; Cardiovascular Disease; Hyperlipidemia; Inflammation; Diabetes; Obesity; Cell Signaling; AMPK; Mtorc1; PI3K; FOXO
Abbreviations
ACC: Acetyl-CoA Carboxylase; ACE: Angiotensin Converting Enzyme; AF: Atrial Fibrillation; AGE: Advanced Glycation End- Product; Aκt: Protein Kinase B; AMPK: AMP-Activated Protein Kinase; ANGPTL: Angiopoietin-Like; AP-1: Activated Protein-1; APC: Activated Protein C; Apo: Apolipoprotein; aPTT: Activated Partial Thrombin Time; AT III: Antithrombin; AT: Angiotensin; AXL: Receptor Tyrosine Kinase AXL (Anexelekto; Uncontrolled); BAS: Bile Acid Sequestrants; BP: Blood Pressure; C/REBP: cAMP Response Element-Binding Protein; cAMP: Cyclic Adenosine Monophosphate; cGMP: Cyclic Guanosine Monophosphate; CM: Chylomicron; COX: Cyclooxygenase; CRP: C-Reactive Protein; CVD: Cardiovascular Disease; DHA: Docosahexaenoic Acid; DPP- 4: Dipeptidyl Peptidase 4; EC: Endothelial Cell; ECM: Extracellular Matrix; EGC: Epigallocatechin; EGCG: EGC Gallates; EPA: Eicosapentaenoic Acid; ERK: Extracellular Signal Regulated Kinase; ET: Endothelin; FAS: Fatty Acid Synthase; FBG: Fibrinogen; FH: Familial Hypercholesterolemia; FIIa: Thrombin; FOXO: Forkhead Box O; GAS6: Growth Arrest-Specific 6 Protein; GC: Gallocatechin; GI: Gastrointestine; GLP-1: Glucogan-Like Protein-1; GP: Glycoprotein; GPIHBP1: Glycosylphosphatidylinositol-Anchored High-Density Lipoprotein Binding Protein 1; GPx1: Glutathione Peroxidase 1; GSH: Reduced Glutathione; GSK3ß: Glycogen Synthase Kinase 3ß; Hb: Hemoglobin; HDL: High Density Lipoprotein; HDL-C: HDLCholesterol; HF: Heart Failure; HIF: Hypoxia Inducible Factor; HMGB1: High Mobility Group Box 1; HO-1: Heme Oxygenase-1; HSL: Hormone Sensitive Lipase; HSYA: Hydroxysafflor Yellow A; Hyper TG: Hypertriglyceridemia; I/R: Ischemia/Reperfusion; Idol: Inducible Degrader Of LDLR; IFN: Interferon; IκB: Inhibitor Kappa B; IKK: IκB Kinase; IL: Interleukin; iNOS: Inducible NOS; IRS: Insulin Receptor Substrate; IsoP: Isoprostane; JAK: Janus Kinase; JNK: Jun N-Terminus Kinase; LDL: Low Density Lipoprotein; LDL-C: LDL-Cholesterol; LDLR: LDL Receptor; LMWH: Low- Molecular-Weight Heparin; Lp[a]: Lipoprotein [a]; LPL: Lipoprotein Lipase; LV: Left Ventricular; LX: Lipoxin; mAB: Monoclonal Antibody; MAPK: Mitogen-Activated Protein Kinase; MCP-1: Monocyte Chemoattractant Protein 1; MI: Myocardial Infarction; miR: MicroRNA; MMP: Matrix Metalloproteases; mTORC: Mammalian/Mechanistic Target Of Rapamycin Complex; MTP: Microsomal Triglyceride Transfer Protein; MF: Macrophage ; NFAT: Nuclear Factor Activated T; NFκB: Nuclear Factor Kappa B; NLRP: NOD-Like Receptor Protein; NOS: Nitric Oxide Synthase; NOX: NADPH Oxidase; Nrf2: Nuclear Factor Erythroid 2-Related Factor 2; NSAID: Non-Steroid Anti-Inflammatory Drug; NT-ProBNP: N-Terminal Pro–Brain Natriuretic Peptide; OxLDL: Oxidized LDL; PAF: Platelet Activating Factor; PAI: Plasminogen Activator Inhibitor; PAR: Protease-Activated Receptor; PCSK: Proprotein Convertase Subtilisin Kexin; PDE: Phosphate Diesterase; PGC-1a: Peroxisome Proliferator-Activated Receptor Coactivator; PGE2: Prostaglandin E2; PGI2: Prostacyclin; PI3K: Phosphatidylinositol 3-Kinase; PPAR: Peroxisome Proliferator-Activated Receptor; PT: Partial Thrombin Time; PTEN: Phosphatase and Tensin Homolog; RAAS: Rennin-Angiotensin-Aldosterone-System; RCT: Reverse Cholesterol Transport; ROS: Reactive Oxygen Species; Rv: Resolving; SCFA: Short Chain Fatty Acid; sGC: Soluble Guanylate Cyclase; SirT: Sirtuins; SOD: Superoxide Dismutase; SREBP: Sterol Response Element Binding Protein; STAT: Signal Transducer and Activator of Transcription; SVEP1: Sushi, Von Willebrand Factor Type A, EGF and Pentraxin Domain Containing 1; TAFI: Thrombin Activatable Fibrinolysis Inhibitor; TF: Tissue Factor ; TFPI: TF Pathway Inhibitor; TG: Triglyceride ; TLR: Toll-Like Receptor ; TMA: Trimethylamine; tPA: Tissue Plasminogen Activator; Treg: Regulatory T Cells; TSC: Tuberous Sclerosis Complex; TT: Thrombin Time; TxA2: Thromboxane A2; UCP1: Uncoupling Protein 1; VLDL: Very Low Density Lipoprotein; VSMC: Vascular Smooth Muscle Cell; vWF: Von Willebrand Factor
Introduction
In retrospect, the twentieth century marked cardiovascular disease (CVD) as the most common mortality in US, which reached a peak-high death rate of nearly 350 deaths per 100,000 populations around 1950s to 1970s followed by progressive modest reductions. A meta-analysis by the CDC has reported nearly 45% falling deaths from CVD (e.g., myocardial infarction (MI), heart failure (HF), unstable/ chronic angina) in US between 1980 and 2000 [1], which is followed by an unchanging/flattening trend of cardiovascular mortality thereafter. The cardioprotection has been almost equally attributed to pharmaceutical treatments (47%) and risk-factor reductions (44%) [1]. The treatments have resulted from resuscitation, thrombolysis, aspirin, statins, ß- blockers, ACE inhibitors, warfarin, etc. while lifestyle changes have involved initial and primary cardioprotection. The reduced prevalence of major CVD a risk has included reductions in total cholesterol, systolic blood pressure, smoking, and physical inactivity. However, increases in MBI and diabetes have hiked the deaths by 8% and 10%, respectively.
Similarly, National Health and Nutrition Examination Survey [2] revealed significant decreases in overall prevalence of coronary heart disease (CHD) from 10.3% to 8.0% in the US population between 2001 and 2012 among aged >40 years, which included angina and MI declines from 7.8% to 5.5% and from 5.5% to 4.7%, respectively.
It is also proposed that a healthy lifestyle (no current smoking, no obesity, regular physical activity, and a healthy diet) could offset an elevated genetic risk for coronary artery disease. Diet is one of modifiable CVD risk factors; it becomes the first consideration for cardiovascular health. Accordingly, dietary therapy is the first line prior to any medication. Typical nutritional modifications of CVD risk factors could involve enhanced endothelial NO production (by arginine, antioxidants: CoQ10, lipoic acid, vitamin C/E, glutathione, and eNOS cofactors: B2, B3, BH4, folate), protection from LDL oxidation (by antioxidants, vitamin C/E, ß-carotene), lipid lowering (by conjugated linoleic acid, n-3 FAs), and lowered plasma homocysteine (by B6, B12, folate) [3]. For instance, B3 not only increases HDL-C by 30%, but also significantly lowers lipoprotein [a] (Lp[a]).
The French paradox certainly underscores the benefits of phytochemicals (e.g., polyphenols) in cardioprotection; ever since, it has surged in-depth basic research and clinical trials for diverse disease prevention and intervention beyond CVD including cancers, diabetes, neurodegenerative and inflammatory diseases, etc. This review briefly summaries major CVD types, risks, and typical pharmacological treatments followed by reviewing polyphenols, a significant group of bioactive compounds in phytochemical superfamily, that multiply target CVD risks, readily conferring broad cardioprotection and benefits to CVD.
Common CVD
CVD, a non-communicable disease, presents a group of disorders of the heart (e.g., HF, MI, hypertrophy, arrhythmia including atrial fibrillation (AF), etc.) and blood vessels (vascular diseases: e.g., atherosclerosis, hypertension, and thrombosis). HF, cardiomyopathy, and cardiac arrhythmia often involve increased [Ca+2]i and abnormal myocyte Ca+2 signaling, while cardiomyocytes apoptosis mediates HF. Lack of cardiac energy involving defects in substrate (e.g., fatty acid, glucose) utilization, mitochondrial oxidative phosphorylation, and ATP transfer also plays a contributing role, being recognized as a chemical nature of HF. The interplays among different major CVD types (atherosclerosis, MI, cardiac hypertrophy, arrhythmia, AF, HF) forming feed-forward loops make CVD so complicated. As a metabolic syndrome, CVD significantly overlaps with other members including diabetes, obesity, and non-alcoholic fatty liver disease, exhibiting diverse risks and complexity.
Atherosclerosis
Atherosclerosis is a disease of the large arteries, which is a major cause of CVD conferring HF. It is characterized by the accumulation of cholesteryl esters, microphages (MF), and fibrous elements in the intima. The rupture of such lesions can result in MI and the formation of thrombi, which in turn leads to HF. Apart from the lipid hypothesis of cholesterol accumulation, atherogenic risks include oxidative stress, infection, inflammation, shear stress, endothelial dysfunction, homocysteine, diabetes, obesity, and genetic factors.
It has long been established that there are three distinct phases for atherogenesis: fatty streak formation and fibrous cap formation followed by plaque rupture. Vascular cells (monocytes, VECs, VSMCs, platelets, etc.) and immune cells (e.g., MFs, leukocytes, neutrophils, mast cells, DCs, T/B lymphocytes, etc.) all participate in atherogenic progression [4]. Initially, circulating monocytes enter intima and differentiate into MFs. MF proliferation and accumulation takes up cholesterol/OxLDL-C to form foam cells within the lesion, playing a major role in progression and worsening of atherosclerosis. MF colony stimulating factor likely promotes such MF proliferation and accumulation; MFs play significant roles in atherosclerosis severity and its progression into MI and HF. While MF cholesterol efflux could lead to regression of plaque formation, MF apoptosis decreases collagen synthesis (VSMC apoptosis) and thins fibrous cap, triggering rupture and thrombosis. It is proposed that MF retention in the lesions favors atherogenic progression [5]. Furthermore, MF polarization also plays important roles [6]. For instance, M0 MFs express CD163 (a hemoglobin (Hb) receptor) leading to heme oxygenase-1 (HO-1) activation, while CD36/SR-A expression is responsible for OxLDL uptake. M1-derived MMP1/3/9 promotes matrix remodeling, fibrous cap thinning, and plaque rupture. M2-derived cytokines (IL-1/4/13/10) and vitamin D reduce EC activation via their antiinflammatory effects. The cytokines promote VSMC activation/proliferation, and favor Th2 and Treg development. Moreover, M2 MFs are responsible for suppressed foam cell formation, reduced plaque cholesterol uptake, and reverse cholesterol transport (RCT) as well as wound repair and tissue remodeling. M4 MFs lead to EC activation/dysfunction as well as VSMC activation without enough OxLDL and Hb uptake, correlating to plaque instability/vulnerability.
VSMC proliferation in response to cytokines (e.g., TNF, ILs), adhesion molecules (e.g., VCAM-1, ICAM-1), growth factors (e.g., PDGF) in concert with collagen formation and MMPs play central events in the 2nd phase of fibrous cap formation. VSMC migration into the neointima and its interaction with EC at lesion-prone sites might trigger an inflammatory response in the vessel wall early in the genesis of atherosclerosis and contribute to destabilization of advanced atherosclerotic lesions. Atherosclerosis could continually progress into MI following lesion rupture of the phase III.
In summary, atherogenesis is promoted by decreased NO, increased adhesion molecules (e.g., VCAM-1, ICAM-1), cytokines (e.g., TNF, IL-1), oxidative stress, growth factors (e.g., PDGF), MMP, and ET-1.
Myocardial infarction
Occluded artery per se results in insufficient oxygen supply (ischemia) to myocardium, changing cellular and extracellular components and manifesting at the tissue level of altered wall structure, chamber geometry, and pump function. In this regard, ROS essentially plays an important role in MI development [7] following ischemia injury. Type 1 MI occurs with coronary thrombosis, whereas Type 2 MI with high mortality results from myocardial ischemia. Subsequent re-introduction of oxygen (reperfusion), however, leads to extensive membrane damage and apoptotic or necrotic tissue death during cardiac infarction, manifesting as profound consequence. In addition, MI is the most commom major vascular complication after non-cardiac surgery that often leads to platelet activation for thrombus formation.
In post-MI, inflammatory phase (e.g., MF and neutrophil infiltration) initiates wound healing (e.g., fibroblast and EC activation) and scarring (e.g., fibrosis; excessive ECM accumulation). Stable scar and adverse remodeling could contract heart muscle and lead to congestive HF. Following MI, peripheral blood monocytes in response to chemotactic factors (e.g., MCP-1) migrate into infarcted myocardium and differentiate into MFs that play major roles in phagocytosis (e.g., necrotic myocytes), efferocytosis (e.g., apoptotic neutrophils), cytokine/chemokine/growth factor production, and angiogenesis. Similar to atherogenic proceeding, MFs play dictating roles in post-MI. Depending on MF polarization, proinflammatory
M1 MFs favor inflammation which drives wound healing and ECM destruction (e.g., MMP-9 production); whereas, antiinflammatory M2 MFs prefer ECM reconstruction and angiogenesis. M1 and M2 polarization are reversible and mutually suppress each other.
Thus, MFs become targets for determining MI outcomes. Diverse MF cytokines/chemokines/growth factors/angiogenic molecules (e.g., TGFß1) activate cardiac fibroblasts into myofibroblasts that pave the ways to either wound healing or scarring (fibrosis). Myofibroblasts drive aggressive remodeling of the ECM and wound contraction, enabling rapid and effective repair of the cardiac interstitium [8] for determining MI outcomes. Furthermore, MF-derived angiotensin converting enzyme (ACE) could involve the risk of recurrent MI with left ventricular (LV) dysfunction.
Cardiac hypertrophy
Cardiac hypertrophy (increase in size with diminished contractile), especially LV hypertrophy, significantly contributes to HF. Although exercise, pregnancy, or even big meals could lead to physiological hypertrophy of normal cardiac enlargement/ remodeling, pathological hypertrophy is strictly attributed to stresses (e.g., oxidative stress, inflammation, hypertension, myocardial injury, neuro-activation/stimulation, etc.).
Either activation of PI3K/AkT/mTOR signaling by insulin/ adipokine or PPAR complex (PGC1a and RXR) upregulation on transcription and protein synthesis by high fat (especially saturated) could mediate hypertrophy in the context of the hypertrophic pathogenesis of cardiac growth as the consequence of enhanced protein synthesis. Specifically, enhanced protein synthesis results from nuclear signaling involving ß-catenin/Wnt-dependent or calcineurin/nuclear factor activated T cell (NFAT) pathways. For instance, endothelial dysfunction with AT-II or ET-1 elevation induces increase in [Ca+2]i to activate calcineurin (PP2B) that dephosphorylates NFAT for its nuclear import proceeding with gene upregulation, thus manifesting as hypertrophy. Similarly, nuclear import of catenin results in hypertrophy. Pressure overload-mediated Notch signaling is also proposed to lead to cardiac hypertrophy [9]. In contrast, FOXO nuclear import promotes atrophic gene (atrogene) expression (e.g., atrogin-1), thereby repressing cardiac growth. AkT phosphorylates FOXO for nuclear exclusion to dampen FOXO antihypertrophic action [10,11]. Class II or class I DHAC respectively turns on or off hypertrophic gene expression [12].
Regarding to inflammatory responses such as MEK/ERK activation [13] or TNF-mediated ROS [14], the resulting NFkB activation triggers hypertrophic gene expression. Similarly, ROS mediates Ras/Rac effect on hypertrophy. As a downstream event of hypertension, endothelial dysfunction in response to elevated ATII, ET-1, or homocysteine also involving ROS plays a contributing role. Accordingly, AT-II also leads to the development of myocardial hypertrophy. So does reduced NO in endothelial dysfunction per se promotes such; NO activates soluble gaunylate cyclase (sGC) for cGMP formation and cGMP and PKG are negative regulators for cardio-hypertrophy. cGMP causes Ca+2 effluxes, resulting in VSMC relaxation, while PKG phosphorylates myosin phosphatase to actually dephosphorylate myosin, promoting contractile. In addition, catecholamine induces myocardial hypertrophy. Biochemically, alteration in membrane lipid composition (e.g., PIP2) of cardiac myocytes could participate in cardiac hypertrophy, cardiomyopathy, and infarction; PIP2 could stimulate inward racfying K+ channel with enhanced conduction due to favored intercellular coupling. By activating TNF-a -associated calcineurin-NFAT signaling, TxA2 mediates iron-overload cardiomyopathy.
Arrhythmia
Arrhythmia often presents as atrial and ventricular fibrillation with abnormal electrical activity (either individual cell’s electrophysiology or cell-to-cell propagation) of the heart recorded on electrocardiogram. Arrhythmia could result from abnormalities in impulse initiation (triggered activity, automaticity) and conduction (reentry) with a wide variety of abnormalities including myocardial scar, atrial fibosis, adrenergic surge, inflammation, acute ischemia, wall tension due to stretch and drug reactions, genetic factors, etc. Disrupted ion channel activities appear to be the common mechanistic contributor to arrhythmia or even arrhythmic sudden death (e.g., Na+ channel). Arrhythmia (e.g., AF) and HF are mutual cause or result for each other. AF often occurs in association with acquired diseases such as hypertension and valvular heart disease. Arrhythmia also often complicates MI, while AF appreciably increases risks for stroke and HF.
Mitochondrial dysfunction with impaired intracellular ion homeostasis could adversely affect cardiac electrical function, while reduced ATP production and excessive ROS generation could result in increased propensity to cardiac arrhythmias [15]. Catecholamine is known to induce arrhythmias. Mutations in ryanodine receptor modulating [Ca+2] i also trigger cardiac arrhythmias.
AF is the most common sustained cardiac arrhythmia, which is attributed to atrial structural remodeling (e.g., fibrosis) associated with congestive HF. NADPH oxidase (NOX) plays a pathogenic role in AF [16,17]. Left atrial fibrosis is prominent in AF. Atrial fibrosis increases vulnerability to AF, involving elevated AT-II with increased ERK activation and overexpression of atrial TGFß1. While alcohol consumption posing AF risk, numerous genetic factors contribute to AF pathogenesis involving subunits of K+ or Na+ channels, sarcolipin gene, (RAAS) gene, connexin-40 gene, eNOS gene, and IL-10 gene.
Heart failure
HF characterized with heart chamber dilatation and contractile dysfunction fails to supply enough blood to tissues manifesting severe fatigue, shortness of breath, fluid retention, and ultimately multiple organ failures and death. Many late-stage HFs manifest as arrhythmia (abnormal heart rhythm) and sudden cardiac death.
HF is heterogeneous [18] resulting from lack of cardiac energy, ischemia/reperfusion (I/R), myocardial cell apoptosis/cell death/ necrosis, cardiac hypertrophy of sustained overload, enhanced fibrosis, and/or defect in contractile machinery (Ca+2 cycling of impaired Ca+2 homeostasis), most of which are also of predisposition among each others. HF also generally involves hemodynamic as well as neurohormonal components such as elevated levels of N-terminal pro– brain natriuretic peptide (NT-proBNP) and cardiac troponin in acute HF. A variety of neuro-hormones (e.g., renin, angiotensin (AT), aldosterone, etc.) readily induce HF. Moreover, phosphate diesterase (PDE) degrades cGMP, which could lead to chronic HF; PDE9 and PDE5, respectively, degrade ANP- or BNP-formed and NO-derived cGMP. Of particular interest, anemia (hemoglobin <10 mg/dl) is prevalent in HF population, showing enhanced mortality.
Major CVD Risks
The classical CVD risk factors include hyperlipidemia, hypertension, and obesity/diabetes. The newer risk factors for instance include homocysteine, fibrinogen (FBG), impaired fibrinolysis, increased platelet reactivity, hypercoagulability, Lp[a], small dense low-density lipoprotein cholesterol (LDL-C), and inflammatoryinfectious markers. In recent developments, the common causal risk factors for CHD include BMI, LDL-C, TG, IL-6R, Lp[a], and IL-1. In contrast, C-reactive protein (CRP), homocysteine, HDL-C, and lipoprotein-associated phospholipase A2 are not strongly included as factors, since HDL-C increases and phospholipase A2 inhibition do not significantly correlate to CVD reduction.
In another categorization, modifiable CVD risks include healthy diet, obesity, smoking, physical activity, hyperlipidemia, elevated blood pressure, metabolic syndrome, or diabetes mellitus. By contrast, aging, ethnicity, gender, blood type, and genetics are not modifiable. Male gender is generally more susceptible, but postmenopausal women have enhanced risk than men.
Major CVD risks are summarized in Figure 1 (left panel). Among which, interactions exist among risks such as oxidative stress/ inflammation axis, inflammation/thrombosis loops, and obesity/ diabetes cross-talks, all driving CVD progression. As part of metabolic syndrome, CVD significantly overlaps with other members including diabetes, obesity, and non-alcoholic fatty liver disease. The genetic factors often complicate CVD risks (please refer to [19,20]). Genetic testing is highly recommended, which prompts earlier prevention including those modifiable risks such as lifestyle improvements.
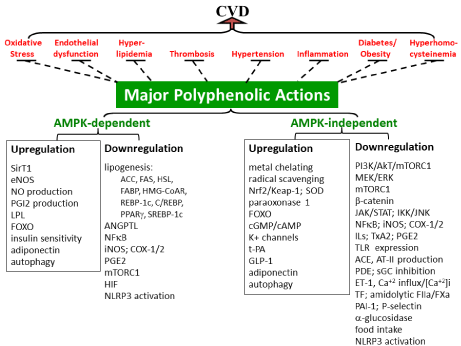
Figure 1: Multiple targets of CVD risks antagonized by polyphenols. The left panel depicts major risks (key characteristics in parentheses) for contributing to CVD.
The right panellists’ diverse polyphenolic actions that multiply target common CVD risks (left panel); major events in each functional category are highlighted in
parentheses. Blunted dash-lines indicate antagonisms; please refer to the text for details. ↑ denotes upregulation, activation, or increased production, while ↓
denotes downregulation, inhibition, inactivation, or suppressed expression/production.
Risk assessments and predications
There are three major models assessing risks for CVD (MI, coronary heart disease (CHD), stroke, and transient ischemic attacks). (1) The classical US Framingham model developed since 1991 does not include measurement on social deprivation, family history, BMI, or current treatment with antihypertensives. (2) Scottish Heart Health as the ASSIGN model based on Framingham adds social deprivation and family history. (3) The QRISK (an algorithm in UK used by British National Health Service) estimates and predicts risks for MI, CHD, stroke, and transient ischaemic attacks. Risk factors include age, sex, smoking status, systolic blood pressure, ratio of total serum cholesterol to HDL-C, left ventricular hypertrophy, BMI, family history of CHD in first degree relative aged less than 60, area measure of social deprivation, and existing treatment with antihypertensive agent (e.g., thiazide, ß blocker, calcium channel blocker, or ACE inhibitor). Apparently, QRISK is more discriminative than Framingham model/algorithm, which however presents better the assessment advantages (nearly closed 1 predicated/observed ratios in CVD risks for 10-year duration), although all three well predict CVD risks compatibly.
Recently, QRISK3 is even more discriminative including more risk factors: age, ethnicity, deprivation, systolic blood pressure, BMI, total-to-HDL cholesterol ratio, smoking, coronary heart disease in a first-degree relative younger than 60, presence of diabetes I/II, treated hypertension, rheumatoid arthritis, atrial fibrillation, and stage 4 or 5 chronic kidney disease as well as new independent CVD risk factors (e.g., stage 3 chronic kidney disease, systolic blood pressure variability, migraine, corticosteroid use, systemic lupus, atypical antipsychotic use, severe mental illness, and erectile dysfunction). It is expected to achieve a precise CVD risk predication.
Oxidative stress
Biological system is constantly under oxidative stress, not only living in 20% oxygen (O2) atmosphere, but also hypoxia (ischemia) stabilizing HIF1a to upregulate NADPH oxidase (NOX) (superoxide anion (O2-), formation) or to turn on downstream angiogenic gene (e.g., VEGF) expression. Oxidative stress serves as a molecular mechanism to mediate diverse disease progression and pathogenesis. In a classical view of singlet O2 metabolism, molecular O2 is utilized by biological systems followed by a consequence of formations of O2-, hydrogen peroxide (H2O2), hydroxyl radical (OH), and H2O in stepwise one-electron sequential reductions [21]. O2 -, H2O2, and OH are three major reactive oxygen (ROS), all of which are cytotoxic and exhibit damaging effects on biological components including DNA damage, lipid/cholesterol oxidation, lipoprotein oxidation, protein oxidation, and membrane disruption [22].
The heart is a highly oxidative tissue with an oxygen utilization rate of 60-150 mmol/min in humans; over 90% of heart metabolism is aerobic. Heart tissue is also remarkably sensitive to oxygen deprivation (ischemia). Vascular cells are also susceptible to oxidative stress. Oxidative stress could trigger atherosclerosis, thrombosis, hypertension, hyprertrophy, and arrhythmia. In addition to initiating inflammatory atherosclerosis, oxidized LDL (OxLDL), for instance, drives platelet activation/aggregation by stimulating NOX and O2- production that in turn suppresses NO/cGMP/PKG pathway.
In triggering CVD, ROS has been proposed to mediate arrhythmia; NOX plays a role in AF. ROS activates NFkB and favors hypertrophic gene program. (1) ROS is a known factor for endothelial dysfunction, a key event for CVD risk (see next section on endothelial dysfunction). For instance, O2- limits the biological activity of NO by OONO- formation; as a result, VSMC proliferation is encouraged as an essential component in atherogenesis. (2) ROS modulates coagulation pathway, fibrinolysis, and platelet aggregation. Excessive vascular O2•- and H2O2 promote platelet activation/recruitment and thrombus formation; oxidation readily upregulates coagulation and vWF binding to platelets, favoring thrombosis. There is evidence that protein methionine oxidations by H2O2, HOCl, and other ROS form methionine sulfone, which could play pathogenic roles in atherosclerosis, ischemic heart disease, hypertension, and thrombosis. (a) ROS can promote the initiation of coagulation by targeting the tissue factor (TF)-FVII complex. Oxidized lipids also promote formation of procoagulant TF microparticles derived from monocytes. Methionine oxidation to methionine sulfoxide also upregulates coagulation factors.
(b) Increased OxLDL or oxidized PLs as platelet CD36 ligand activates MAPK (ERK5; a redox sensor) for ROS production including O2•- and H2O2, which promotes platelet activation and thrombus formation. (c) While enhancing PAI-1 activity, ROS also inhibits the production of activated protein C (APC), thus favoring coagulation and formation of thrombin and thrombus.
(d) The oxidative product methionine sulfone(s) could alter protein functions and enzyme activities for suppressing apolipoprotein (Apo) A-I, actin, p53, S100A9, thrombomodulin, APC, ADAMTS13, and clotting factor VII (FVII) while enhancing CaMKII, IkB , and von Willebrand factor (vWF), thereby all contributing to thrombotic vascular diseases and CVD. Methionine sulfoxide increases the ability of VWF to tether platelets; the oxidation essentially destabilizes the A2 domain for resistance to proteolysis by ADAMTS13; thus, this relatively increases/favors A1 ability to bind platelet receptor GpIba for platelet activation/ aggregation for thrombus formation.
(c) O2•- drives further platelet activation and recruitment leading to greater thrombus formation.
(e) H2O2 promotes phosphorylation of AXL in VSMCs, while ATII and thrombin increase AXL expression in VSMCs. (i) Endothelial AXL essentially functions as a phagocytic receptor. GAS6-AXL protein interaction mediates endothelial uptake of platelet microparticles, which is mediated by exposed PS on microparticles along with induced ICAM and E-selectin expression, presenting thrombotic events. (ii) GAS6/AXL/AkT pathway tyrosine phosphorylates aIIbß3 integrins, which enhances platelet outsidein signaling promoting platelet aggregation for thrombosis (3). Following free radical reactions under oxidative stress, arachidonic acid (AA)-derived 15-F2t- isoprostane (IsoP) or 15-E2t-IsoP mediated by TxA2 receptor exhibits bronchoconstriction, vasoconstriction, platelet aggregation, and adhesion. Similarly, 15-F2c-IsoP activates PGF2 receptor and induces hypertrophy in cardiac smooth muscle cells, while Isoketals-protein adducts present in atherosclerosis and MI (4). Redox-inflammation axis certainly poses CVD risks. ROS cytotoxicity triggers inflammatory responses via either intra- or extra- cellular signaling [23,24] involving HIF stabilization [25,26], IKK/JNK upregulation, STAT3 activation, NFkB/AP-1 activation, COX-2 activation, inducible nitric oxide synthase (iNOS) activation, and cytokine production.
Intrinsic ROS sources include by-products of mitochondrial respiration, peroxisomal enzymatic reactions (peroxisomal oxidases, acyl-CoA oxidase, xanthine oxidase), innate immunity of respiratory burst (NOX activation) during infection, non-enzymatic Fenton reaction upon Fe(II) Oxidation to Fe (III), and in response to endogenous homocysteine [27], ET-1 [28], AT II [29,30], OxLDL, and AGEs [31] (see the below section on Endothelial dysfunction). Interestingly, OxLDL activates NOX, refueling oxidative stress. Smoking, alcohol, xenobiotic oxidation by cytochrome P450, and UV/radiation contribute to major extrinsic ROS.
Hyperlipidemia
Among the classical Frederickson’s classifications, all six phenotypes (Type I, IIa, IIb, III, VI, and V) result from differential hyperlipidemic mechanisms involving either genetic or acquired factors. Type I is primarily characterized by lipoprotein lipase (LPL) and ApoC-2 deficiencies with elevated chylomicrons (CM). Type IIa, IIb, and IV are familial combined hyperlipidemia characterized by elevated plasma cholesterol and triglyceride (TG) due to increases in LDL and VLDL production. Type III shows accumulation of CM and VLDL remnants, largely resulting from hepatic lipase deficiency and ApoE mutation/deficiency as receptor defects. Type IV is characterized by elevated plasma TG and increased VLDL. The mixed type V is characterized by elevated plasma TG and cholesterol with increased VLDL and CM due to their overproduction and defective clearance.
Biochemically, LDL is primarily responsible for cholesterol forward delivery and prothrombotic actions, while HDL plays pivotal roles in RCT process as well as antithrombotic actions including attenuated vWF-dependent/initiated platelet adhesion, megakaryocyte progenitor cell proliferation, and platelet production. In hyperlipidemia, increased OxLDL or oxidized PLs as platelet CD36 ligand activates MAPK (ERK5; a redox sensor) for ROS production including O2•- and H2O2, which promotes platelet activation and thrombus formation. Oxidized lipids also promote formation of procoagulant TF microparticles derived from monocytes. LPL expression and secretion are subject to regulation of plasma TG: LPL deficiency becomes a genetic detrimental factor for hyperTG.
(1)Hypercholesterolemia: Nearly 80% of plasma cholesterol derives from de novo biosynthesis in the liver; other 20% or so is attributed to dietary intake. Intracellular cholesterol pool essentially dictates cholesterol homeostasis. Upregulated cholesterol biosynthesis and high fat intake with high cholesterol content readily contribute to atherogenic risk. The efficiency of LDL-C uptake by hepatic LDL receptor (LDLR) plays a central and dictating role in hypercholesterolemia, which concerns cholesterol homeostasis including LDLR expression/ recycling, internalization mediated by proprotein convertase subtilisin kexin-9 (PCSK-9), and lysosomal degradation by inducible degrader of LDLR (Idol). Interestingly, variants in HMG-CoA reductase also play significiant roles in such cholesterol homeostasis. Biochemically, (a) involving in either genetic or acquired factors, hypercholesterolemia is predominant in CVD, especially atherosclerosis that is characterized by cholesterol accumulation in intima, thereby narrowing vascular lumen and limiting blood flow; (b) apart from the lipid hypothesis, hypercholesterolemia (i) mediates platelet production and activation readily triggering atherosclerosis, (ii) could refer to a proinflammatory state in view of intracellular cholesterol accumulation (foam cells, SMCs, and other immune cells in intima) surging cytokine production for atherogenic progression; cholesterol crystals readily involve in NLPR3 inflammasome activation and atherogenesis [32], and (iii) promotes IFN release from T cells and CD40/CD40L [33] response for activating NOX, both of which lead to vascular inflammation.
Genetically with defects/deficiencies in LDLR pathway, homozygous familial hypercholesterolemia (FH) presents LDL-C in the range of 500 to 1000 mg per deciliter and severe premature atherosclerosis, while LDL-C in heterozygous FH is typically well above the 95th percentile according to age and sex bases [19,20].
Thus, LDL-C is not only a risk factor but also a biomarker for atherosclerosis. The current guideline sets LDL-C goals at 70, 100, 130, and 160 mg/dl, highly depending on other existing risk factors (e.g., degrees and durations) according to the National Cholesterol Education Program Adult Treatment Panel III (2004) by NIH-the National Heart, Lung, and Blood Institutes. The recent recommendations/modifications by the American College of Cardiology/the American Heart Association further include treatment with statin and its dosages [34].
(2)Hypertriglyceridemia: Hypertriglyceridemia (hyperTG) defines plasma TG level greater than 200 mg/dl and the plasma concentrations can exceed 1,000 mg/dl in severe cases. Plasma TG levels (≥ 500 mg/dL) shows a significantly lower survival rate than those with “low-normal” levels of <100 mg/dL. Even high-normal TG levels (100-149 mg/dL) are associated with increased mortality.
The level of plasma TG is, in part, heritable. TG-rich lipoproteins generally reflect plasma TG level. The etiology also includes highcarbohydrate diet, high-fat diet, obesity, and renal failure, which could also be associated with diabetes, alcohol abuse, nephrotic syndrome, hypothyroidism, autoimmune disorders, paraproteinemias, and pregnancy. Some prescribed medications (e.g., glucocorticoids, estrogens, tamoxifen, hydrochlorothiazide, non-selective ß-blockers, clozapine, and lanzapine) are known to increase TG levels. HyperTG accompanied by elevated TG-rich lipoproteins (e.g., VLDL, LDL) is associated with atherosclerosis, stroke, and insulin resistance, thereby increasing the risk for coronary artery disease [35-37]. For instance, acute hyperTG induces platelet hyperactivity [38], while elevated VLDL promotes PAI-1 gene expression [39], favoring thrombus formation. Beyond CVD, severe hyperTG can have serious medical consequences such as acute pancreatitis.
(a) Role of LPL. LPL, synthesized in cells from adipose and skeletal muscle, is scereted into the subendothelial space and transported to the luminal surface of capillary ECs. In capillaries, ECs express glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1) that binds LPL. GPIHBP1 facilitates LPL activity mainly responsible for digestion/hydrolysis of TG-rich lipoproteins (e.g., CMs, VLDL) to CM remnants/IDL and circulating free fatty acids, monoacylglycerol, and glycerol. Thus, LPL activity dictates plasma TG level and genetically determined TG levels lie on LPL mutation as well as mutations of LPL upstream regulators. ApoC1/3 and angiopoietin-like (ANGPTL) 3/4 are known endogenous regulators to inhibit LPL, while ApoC2 and ApoA5 activate LPL. Interestingly, miR-29/410/1277 could also show LPL inhibition. Accordingly, ApoC3 elevation and/or gain-of-function mutation(s) and ApoC2/ApoA5/LPL deficiency or loss-of-function mutation(s) could pose (severe) hyperTG. For instance, APOA5 variants link to the polygenic hyperlipidemia types IIb, III, IV, and V as well as CVD risk. LPL expression and secretion are also subject to regulation of plasma TG. For Instance, vitamin D3 deficiency often suppress LPL expression and deficiency becomes a detrimental factor for hyperTG.
(b) LPL mutation. Loss-of-function mutation (e.g., G161E, Q240H, A98T, L279V, D36N, etc.) leads to LPL deficiency/low activity and elevated TG. In contrast, its gain-of-function mutation (e.g., S447X, S474T, etc.) readily shows protection from hyperTG and against coronary artery disease.
(c) Loss-of-function mutation of positive LPL regulators. Lossof- function of ApoA5 (e.g., S19T, T133R, Q145X, G271C, T1131C, etc.) show LPL deficiency or low activity and increased plasma TG. For instance, ApoA5 (T1131C) correlates to 14% increased risk of coronart artery disease in clinical trials, which is mediated by attenuated LPL and elevated plasma TG. Similarly, ApoC2 (V40T, loss C-terminus, etc.) loss-of-function mutation leads to either ApoC2 deficiency or reduced LPL activity, thereby resulting in elevated plasma TG.
(d) Loss-of-function mutations of negative LPL regulators. Loss-of-function mutations of ApoC1 (e.g., T45S), Apo3 (e.g., A43T, R19X, etc.), ANGPTL3 (e.g., S17X, E129X, K63T, E91G, L164F, Y417C, N147X, etc.), ANGPTL4 (e.g., E40K, G223R, E167K, G77R, etc.) lead to LPL upregulation and reduced plasma TG. For instance, (i) ANGPTL4 (e.g., E40K) results in nearly 35% lower in TG and 50% lower risk of coronary artery disease accompanied by high HDL-C, which is proposed due to ANGPTL4 destabilization; N-terminus of ANGPTL4 is mainly responsible for LPL binding and inhibition. (ii) Loss-of-function of ANGPTL3 similarly decreases plasma levels of TG, LDL-C, and HDL-C as well as enhances insulin sensitivity and decreases serum levels of free fatty acids. (iii) ApoC3 (e.g., A43T) lowers circulating ApoC3, plasma TG, and risk of coronary heart disease. In addition, loss-of-function mutation (D314A) of GALNT2, an ApoC3 upstream modulator, exhibits LPL upregulation and TG reduction; GALNT2 glycosylates ApoC3 into an active protein.
(e) Other genetic factor for hyperTG. Clinically, (i) a new mechanism reveals that GPIHBP1 deficiency shows low plasma LPL, impaired intravascular hydrolysis of TG, and severe hyperTG (e.g., chylomicronemia). GPIHBP1, a GPI-anchored protein in capillary ECs, binds and stabilizes LPL, facilitating LPL lumen transfer and mediating lipolytic hydrolysis of TG-rich lipoproteins. Thus, GPIHBP1 deficicency/mutation/multimerization lose its affinity for LPL, resulting in hyperTG. Consistently, GPIHBP1 autoantibodies found in autoimmune disease (SLE or Sjögren’s syndrome) block the ability of GPIHBP1 to bind and transport LPL, thereby interfering with LPL-mediated processing of TG-rich lipoproteins and causing severe hyperTG. (ii) Mutated SVEP1 (D2702G) contributes to 14% increased risk of coronary artery disease, although its mechanism of action remains largely unclear. SVEP1 is a cell-adhesion molecule acting as a physiological ligand for integrin a9ß1 that plays diverse roles in cell migration, differentiation, triggering tyrosin phosphorylation/signaling, etc.
(3)Elevated Lp[a]. Lp[a] is a well-known independent risk factor for CVD resulting from its procoagulant, prothrombotic, and antifibrinolytic functions. Lp[a] consists of a lipoprotein moiety resembling LDL as well as the plasminogen-related glycoprotein (Apo[a]). Its pathogenic role has traditionally been considered to reflect a dual function of its similarity to LDL causing atherosclerosis, while its similarity to plasminogen causing arterial and venous thrombosis through inhibition of fibrinolysis [40]. Lp[a] consisting LDL moiety is proposed to be internalized through LDL receptor, while Apo[a] per se is uptaken up by plasminogen receptor.
Clinically, packed with proinflammatory oxidized PLs, plasma Lp[a] ranging from 1 to 250 mg/dL and consisting of an LDL-like particle with covalent disulfide bond between Cys4326 of ApoB-100 and Cys4057 of Apo[a], Lp[a] is an independent causal risk factor for MI, stroke, peripheral arterial disease, and calcific aortic valve stenosis. Lp[a] levels are genetically determined, reaching peaked by the age of two and remaining constant throughout the life. Diet and lifestyle changes have little impact on Lp[a] levels.
Endothelial dysfunction
ECs not only structurally function as a liner of blood vessels, but also play important/ dictating roles in maintaining vascular integrity/homeostasis by interacting with a host of vascular cells. Thus, endothelial dysfunction essentially impacts on vascular tone and EC integrity/permeability/function, which in turn leads to detrimental vascular and heart diseases. Both endothelial dysfunction and activation are broadly implicated in vascular diseases (e.g., atherosclerosis, hypertension, etc.) by increasing vasoconstriction, VSMC proliferation, platelet adhesion/aggregation, leukocyte adhesion to EC, LDL oxidation, MMP activation, and plasminogen activator inhibitor (PAI)-1 (a prothombotic factor) production. EC activation results from elevated AGEs and cytokines. Endothelial dysfunction is generally associated with oxidative stress, hypercholesterolemia, infection, inflammation (e.g., sepsis), hypertension, hyperhomocysteinemia, diabetes, and many other risks.
Endothelial dysfunction mainly features elevated AT-II, endothelin (ET)-1, and thromboxane A2 (TxA2) accompanied by reduced NO and prostacyclin (PGI2). AT-II depresses PGI2 and NO production, ensuring endothelial dysfunction. Elevated MMPs destabilize atherosclerotic plaque. Apart from contribution to endothelial dysfunction, ET-1/AT-II/TxA2 elevations and PGI2/ NO reductions in fact are recognized independent CVD risks: NO and PGI2 contributing to vasodilation, while AT-II, ET-1, and TxA2 playing roles in vessel contraction. In addition, TxA2 per se mediates iron-overload cardiomyopathy by activating TNF-a -associated calcineurin-nuclear factor activated T (NFAT) signaling.
(1) Elevated ET-1. ET-1 (a 21-amino acid peptide; potent vasoconstrictor) contributes to the pathogenesis of vascular abnormalities such as hypertension, atherosclerosis, hypertrophy, and restenosis. ET-1 is overexpressed by activated ECs in response to hormones (e.g., adrenaline, AT-II, vasopressin, insulin, or cortisol), peptides (e.g., cytokines, LPS, IL-1, TGF ), ROS (e.g., H2O2), stimuli (e.g., hypoxia, low shear stress, or osmolarity), thrombin, glucose, or OxLDL, all of which upregulate ET-1 promoter gene expression. On the contrary, ET-1 promoter gene expression is negatively regulated by PGI2, NO; natriuretic peptide A/B/C, heparin, PPARγ ligand/activation, and high shear stress for its natural balance of cardioprotection [41]. ET-1 signaling through its GPCRs (ET A/B) largely mediates an increase in [Ca+2]i by increased influx, release from ER stores, and/or mobilization from mitochondria. ET-1 signaling involves activated signaling transduction pathways including the modulation of the adenylate cyclase/cAMP pathway through Gs and Gi (PKA pathway), IP3 activation through Gq/11 activation (PLC pathway), PI3K activation, phosphoinositide pathway, and MAPK activation) [41]. ET-1 also repolarizes potassium currents and electrical conduction via gap junctions, thus increasing Ca+2 influxes. Such [Ca+2]i mobilization is responsible for cellular hypertrophy, growth, cell proliferation and survival angiogenesis, and nociception; for instance, increased [Ca+2]i promotes VSMC proliferation (atherosclerosis) and constriction (hypertension).
(2) Reduced NO and PGI2. In response to elevated AT-II or ET-1, EC activation/dysfunction inhibits eNOS and represses NO availability while increasing MCP-1, chemokine, and adhesion molecule (ICAM, VCAM, and E-selectin) expression [42]. In addition, decreased NO bioavailability as induced by extracellular hemoglobin during hemolysis thereby favors vasoconstriction, endothelial adhesiveness, platelet activation and aggregation, coagulation, and vessel wall cellular proliferation. Endothelium-derived NO has important vasodilator, antiinflammatory, antithrombotic, and growth-suppressing potentials that are relevant to all stages of atherosclerosis. NO decreases [Ca2+]i, glycoprotein (GP) IIB/IIIa expression, TxA2 expression, P-selectin expression, and platelet association with fibrinogen, which accounts for its anti-platelet actions. NO prevents adherence of leukocytes to the endothelial surface and inhibits expression of leukocyte adhesion molecules at the endothelial surface. NO also inhibits ET-1 induced VSMC proliferation and alters expression of noncellular components that constitute the matrix of the vascular wall, making NO relevant to lesion formation, hypertrophy of the vessel wall, and vascular compliance. NO is known to activate sGC, promoting VSMC Ca+2 efflux and relaxation. [42,43]; conversely, AT-II directly inhibits sGC [44], which is interestingly in line with endothelial dysfunction leading to vasoconstriction. ENOSderived NO promotes cardiomyocyte proliferation by inhibiting TIMP-3 expression. S-nitrosylation and inactivation of GRK2 by eNOS-derived NO protect against myocyte death and HF; otherwise, GRK2 phosphorylates ß-androgenic receptor leading to myocyte deaths. Similarly, S-nitrosylation of L-type Ca2+ channels protects from I/R injury. Moreover, PDE degrades cGMP, which could lead to chronic heart failure; ANP- or BNP-formed and NO-derived cGMP are degraded by PDE9 and PDE5, respectively.
PGI2 (prostacyclin) is the primary prostanoid produced by ECs, which plays an important role in vascular homeostasis through its potent vasodilatory and antithrombotic effects. Cardioprotective PGI2, deriving from AA mainly through COX-2, activates adenylate cyclase and elevates cAMP, resulting in an increase in PKA activity for vasorelaxation [42]. Such mechanism also mediates antiinflammation, a cardioprotective action (refer to below section on Inflammation under Major CVD Risks). Furthermore, PGI2 inhibits platelet aggregation and leukocytes adhesion to ECs.
(3) Elevated AT-II. In addition to its pathogenic role in hypertension (refer to below section on Hypertension under Major CVD Risks), diverse AT-II cardiovascular actions [45] include MF stimulation, vascular permeability, endothelial dysfunction, VSMC proliferation and migration, MMP activation, apoptosis, leukocyte recruitment/infiltration, atherosclerotic plaque formation/instability, fibrosis, and thrombosis (e.g., platelet adhesion/aggregation and PAI- 1/2 upregulation) in part mediated by CD40L. Elevated AT-II also leads to the development of myocardial hypertrophy.
Its abilities to activate NOX and downregulate Nrf2 account for oxidative stress [29,30]. AT-II mediating AT1 receptor-dependent Egr-1 transactivation induces PDGF expression that is involved in atherosclerosis and fibrosis. AT-II induces ET-1 production triggering vascular constriction. Oppositing to NO, AT-II directly inhibits sGC [44], which is interestingly consistent with endothelial dysfunction leading to vasoconstriction. AT-II induces vascular dysfunction and inflammation, which even involves immune cell recruitment with increased infiltration of CD11b T bet+ myelomonocytic cells and NK1 1+T bet+ cells into aorta. Consequently, IL-12 expressed by myelomonocytic cells stimulates IFN-γ production by NK cells in vascular wall, driving vacular injury and inflammation.
Thrombosis
There are three major components constituting thrombosis; hypercoagulation results in blood clot overproduction, platelet activation leads to aggregation with plug formation, and fibrinolytic abnormality exhibits the lack/insufficiency of normal clot resolution. Its risk includes hypercoagulable states [46] (e.g., diabetes, obesity, pregnancy, contraceptives, etc.), prothrombotic status (e.g., cancers, antiphospholipid syndrome, heparin-induced thrombocytopenia, etc.), and inherited factors such as deficiencies in antithrombin III, protein C, and protein S as well as FV Leiden, prothrombin gene mutation, and fibrinolytic defects.
Not only as a risk for CVD, does thrombosis per se present a vascular disease with two major forms: venous and arterial thrombosis. Venous thrombosis not only leads to the deadly pulmonary embolism, but also poses a risk for arterial thrombosis in some clinical cases [47]. It is estimated that peripheral arterial disease increases the risk for MI or stroke by 3 times. In fact, thrombosis plays a role in the phase III of atherogenesis. Thrombosis could block blood flow, which limits nutrient and oxygen supplies to cells and tissues, leading to cell death (e.g., heart attack occurring upon cardiac cell deaths). Blood clots noticeably pool in the top right chamber of the failing heart developing dilated cardiomyopathy. Thrombosis also often coexists with myelo-proliferative disorders and hyperhomocysteinemia. Of importance, thrombosis is pro-inflammatory, which forms positive feedback [48] and forward [49] loops in blood coagulationthrombosis- inflammation circuit for posing CVD risk.
ROS is known to modulate coagulation pathway, fibrinolysis, and platelet aggregation (refer to the above Oxidative stress section for details), while hyperhomocysteinemia elevates TF and PAI-1 expression (refer to below section on Hyperhomocysteinemia under Major CVD Risks). Thrombin (FIIa) plays a central role by promoting fibrin production/polymerization and activating platelets and fibrinolytic inhibitors (e.g., prothrombotic PAI-1, and procoagulant TAFI), thereby ensuring propagation of coagulation and thrombosis. Furthermore, FIIa per se via protease-activated receptor (PAR) -2/4 triggers intracellular proinflammatory signaling [48,49]. Other component such as vWF, MMP-2, and hyperlipidemia also promotes thrombosis.
(1) Hypercoagulation. Hypercoagulability [46] presents twofold relevance to CVD risk. (a) Elevated blood coagulation with fibrin overproduction is recognized as an independent risk for atherosclerosis; in fact, TF-initiated extrinsic coagulation readily plays a role in the phase III of atherogenesis. Elevated procoagulant factors (e.g., FVII, FVIII, FX, FBG) and insufficient anticoagulants (e.g., APC, TFPI, and ATIII) all lead to hypercoagulability. In addition, circulating fibrin clots promote platelet aggregation and thrombi formation. (i) ROS induces TF/FVIIa complex and decreases APC production, therefore favoring coagulant proceeding. (ii) Upregulated TF expression initiating the extrinsic coagulation pathway is in response to infection (e.g., bacterial endotoxin, Chlamydia pneumoniae), inflammation (e.g., sepsis, cytokines,), shear stress, hypoxia, OxLDL, Lp[a], homocysteine, phorbol esters, and many others. The activation of intracellular signaling kinases (e.g., PKC, MAPK, PTK) and transcription factors (e.g., NFkB, AP-1, Egr-1) mediates TF expression. (iii) Tissue injury activating protein disulfide isomerase de-encrypts and activates TF. In some cases of intracellular Ca+2 activation, TF function is drastically upregulated without increased TF protein synthesis. (b) Hypercoagulability with elevated active serine protease procoagulants (e.g., FVIIa, FXa, FXIIa, and FIIa) and fibrin overproduction triggers a host of intracellular inflammatory responses [48,49] that could broadly contribute to CVD (refer to below section on Inflammation under Common CVD Risks). FVIIa/FXa/FIIa elicits an array of proinflammatory cytokines through PARs, while FXIIa initiates the formation of inflammatory mediator: bradykinin. Fibrin per se mediated by TLR-4 triggers MyD88/TRIF-dependent signaling of NFkB activation, a hallmark of inflammation [48,49].
(2) Platelet activation/aggregation. (a) In conjunction with activation of the coagulation cascade, thrombus formation involves the adhesion, activation, and aggregation of platelets. (i) Thrombin activates platelets mainly through PARs and GP. PAR-1 is a primary receptor for thrombin by which platelets are activated to aggregate [50]. Platelet aggregation constitutes thrombus formation involving cross-linking of adjacent platelets mediated by the interaction of activated GPIIb/IIIa with distinct amino acid sequences, LGGAKQAGDV and/or RGD, at each end of dimeric FBG molecules [51]. (ii) An alternative pathway describes that thrombin-induced platelet activation results from polymerizing fibrin, which involves the recognition sites in the cross-linking of polymerizing fibrin and surface integrins via GPIb. In fact, GPIb acts as a thrombin-binding site and promotes platelet activation by low thrombin concentrations [52]. (iii) PAR-4-mediated thrombin action promotes Ca+2 influx, ADP release, and TxA2 production in platelets, all of which promote platelet recruitment and activation [53] in addition to leukocyte rolling and adhesion. (b) Either monomeric or polymeric fibrin, a coagulant product, activates GPVI for platelet aggregation and sustains thrombus growth as occlusion. (c) During endothelial dysfunction, COX-1/2 derived-TxA2 activates platelets to induce platelet aggregation and vasoconstriction responsible for atheroscherosis, MI, thrombosis, hypertension, stroke, and cardiac fibrosis. TxA2 inhibits NOS, also mediating platelet aggregation. (d) In endothelial dysfunction, NOX serves as a major EC source of ROS; O2•- facilitates platelet aggregation. Reduced NO favors/augments platelet aggregation, which is the mechanism by which OxLDL triggers platelet aggregation. (e) Platelet activation, aggregation, and plug stabilization are also promoted by either endogenous extracellular ADP mainly deriving from extracellular ATP upon cell apoptosis/ necrosis or collagen exposure following tissue injury. Activated ECs lose expression of CD39 and CD73 and thus also contribute to platelet activation [54]. ADP simultaneously binds to two P2Y receptors (P2Y1 and P2Y12) leading to sustained activation of Rap1b and a conformation change of aIIbß3 integrins from an inactive to an active form; activated aIIbß3 binds FBG to form a platelet aggregate. (f) vWF also dictates thrombosis; N-terminal vWF complexes and stabilizes FVIII, a coagulation factor for promoting blood clotting, while vWF binding to the activated molecular conformation of integrin aIIbß3 (GPIIb-IIIa) essentially mediate platelet aggregation in the presence of FBG and at high shear flow. Upon EC damage, vWF binds to exposed extracellular matrix and changes vWF affinity for the GPIb- IX, thus promoting platelet adhesion, spreading, and aggregation [55]. (g) In hyperlipidemic conditions, scavenger receptor CD36 on platelets recognizes OxLDL during plaque formation in turn promotes thrombus formation. CD36-dependent OxLDL activates MAP kinase ERK5 (a redox sensor) for ROS production including O2•- and H2O2, which promotes platelet activation. (h) MMP-2 enhances platelet aggregation, which is PAR1-dependent. MMP-2 cleaves PAR1 at an extracellular site different from the thrombin cleavage site. Coupling with Gq and G12/13 pathway activation, MMP-2/PAR1 then triggers p38-MAPK phosphorylation, Ca+2 fluxes, and PI3K activation for platelet activation. Platelet Integrin aIIbß3 is a necessary cofactor for PAR1 cleavage by MMP-2.
Interestingly, platelet activation/aggregation per se is inflammatory. Activated platelets undergoing de-granule release an array of cytokines (e.g., IL-1ß/4/8/13/17, TNF, IFNγ , HMBG1), chemokines (e.g., ß-thromboglobulin, platelet factor 4, ENA-78, Groa, RANTES, SDF1a, and P-selectin), growth factors (e.g., EGF, TGF , PDGF AA/AB/BB), CD40/CD40L, and lipid mediators (e.g., TxA2 and PAF) from its granules, all of which contribute to local or systemic inflammation and immune responses. Platelet activation also elevates E-selectin production, ICAM-1 production, and TF expression, forming a forward loop for promoting thrombosis. Complement activation triggered by platelet activation plays a role in CVD pathogenesis [56]. Oppositing to PGI2, COX-1/2 derived- TxA2 by activated platelets induces platelet aggregation and vasoconstriction responsible for atheroscherosis, MI, hypertension, stroke, and cardiac fibrosis in addition to thrombosis. Moreover, TxA2 inhibits NOS, further mediating platelet aggregation. By activating TNF-a -associated calcineurin-NFAT signaling, TxA2 also mediates iron-overload cardiomyopathy.
(3) Hypofibrinolysis. Hypofibrinolysis leads to neointimal disease, thrombi formation, perivascular fibrosis, and MI. (a) insufficient tPA resulting from either genetic or acquired factors leads to suppressed plasmin production; plasmin, an active serine protease, is responsible for cleavage of fibrin clots. (b) Elevated PAI- 1 is responsible for thrombosis, cardiac fibrosis, atherosclerosis, MI. The elevated expression, for instance in response to FIIa [57], TGFß [58] , leptin , ROS, AT-II [60], FFA [61], or CRP [62], inhibits and lowers plasmin formation for dissolving insoluble fibrin clots. PAI- 1 per se is an acute protein and proinflammatory mediator. PAI activity is high in obesity, diabetes, cancers, hyperhomocysteinemia, and aging. (c) Plasma carboxypeptidases recognized as thrombinactivatable fibrinolysis inhibitor (TAFI, a procoagulant factor) attenuates fibrinolysis [63] in favor of fibrin accumulation, playing a role in venous/deep vein thrombosis [64], disseminated intravascular coagulation [65], the acute phase of ischemic stroke [66], and cardiovascular risks [67]. TAFI inhibits various forms of PA-mediated fibrinolysis [68]. TAFI has also been proposed to reduce the ability of fibrin degradation products to protect plasmin from antiplasmin [69]. High TAFI level is reported in diabetes [70], while low rate of TAFI activation is observed in hemophilia A [71].
Hypertension
Not only as a risk for CVD, does hypertension per se present as vascular disease. There is a general perception that hypertension at any age is associated with higher CVD incidence; each 20 mmHg rise in systolic blood pression (BP) and each 10mm Hg rise in diastolic BP could be associated with nearly 60% and 35% higher risks for peripheral artery disease, respectively. As a primary trigger for atherosclerosis, MI, and cardiac hypertrophy leading to arrhythmia and HF, the pathophysiology of hypertension also includes stroke, renal failure, and cognitive impairment beyond CVD concerns. Of importance, chronic hypertension is associated with increased frequency of preeclampsia, placental abruption, fetal growth restriction, preterm birth, cesarean section, and stillbirth. The common risks include aging, race/ethnicity, stress, woman gender (e.g., pregnancy, birth control, and hormone replacement), overweight/obesity, alcohol consumption and smoking, lack of physical activity, high salt, high fat diet, low potassium, and obstructive sleep apnea. The general guideline sets up treatment threshold at systolic 130(140) mm Hg/diastolic 80(90) mm Hg; above which referred to primary hypertension or prehypertension is recommended for initial treatment.
Hypertension largely results from the imbalance between vasodilation and vasoconstriction [72], which is associated with elevated RAAS. Renin, renin receptor, AT-II formation, AT1R, and aldosterone production in the classical RAAS are readily responsible for vasoconstriction; whereas, natriuretic peptides A/B/C, bradykinin, and AT2R endogenously contribute to vasodilation. Natriuretic peptides are capable of lowering blood pressure and preventing sodium retention, while bradykinin is a vasodilator.
AT-II elevation is a typical contributor to hypertension. ATII signal transmission by AT1 but not AT2 receptor mediates vasoconstriction, PAI-1 expression, aldosterone and vasopressin release, central sympathetic activation, cell growth and proliferation; sodium and water retention, inhibited renin release, and endothelial dysfunction, all are characteristics of hypertension. Suppressed VSMC relaxation per se contributes to hypertension under endothelial dysfunction with ET-1 elevation and NO/PGI2 reduction in response to AT-II [73]. While promoting oxidative stress (e.g., NOX activation), AT-II induces ET-1 production triggering vascular constriction; ET-1 involves induced Ca2+ release from intracellular stores, IP3 generation, inhibition of delayed rectifier K+ current, and stimulation of Na+/H+ exchanger. In hypertensive patients, ET-1 is often overexpressed (enhanced ET-1 mRNA); elevated plasma levels of ET-1 have been found in familial hypertension of black population. Concerning intracellular cGMP playing roles in vasodilation, cGMP degradation by PDEs and endothelial dysfunction with reduced eNOS and NO bioavailability also contribute to hypertension. For instance, AA-derived 20-HETE induces hypertension by endothelial dysfunction (elevated ATII & ET-1, attenuated NO, etc.), ACE expression/upregulation, oxidative stress (NOX activation), vascular inflammation/remodeling, vasoconstriction (vascular hypertrophy), HIFa upregulation, VSMC membrane polarization (blocked K+ efflux, increased Ca+2 influx), etc. Interestingly, elevated levels of androgen, both endogenous and exogenous, are correlated with an increase in BP, which is proposed in part by increased 20-HETE generation by CYP4A/4F catalysis; androgen stimulates CYP4A/4F. Testosterone supplementation similarly increases BP; the administration further impairs NO availability.
Hyperhomocysteinemia
Hyperhomocysteinemia has long been recognized as an independent risk factor for atherosclerosis, hypertension, and thrombosis despite several negative clinical trials showing the failure of B-vitamin supplement to ease CVD in the 2000s. Hyperhomocysteinemia is often associated with relative folate, B6, and B12 deficiencies and/or with diabetes [74], aging, male sex, estrogen deficit, renal insufficiency, caffeine, dopamine agonist, and anticonvulsant use. (1) Homocysteine essentially poses oxidative stress (e.g., NOX activation) on cardiovascular system [75], leading to thrombosis (e.g., platelet aggregation), atherosclerosis (e.g., lipid peroxidation, OxLDL, VSMC proliferation), hypertension, hypertrophy, etc. (a) Metabolically, folate/B12 and B6 are involved in removal/conversion of homocysteine to methionine and glutathione, respectively. In line with promoted oxidative stress, homocysteine accumulation is at the expense of reduced GSH formation, an endogenous antioxidant. (b) In addition, impaired glucose-6-phosphate dehydrogenase in hyperhomocysteinemia facilitates oxidative status by inhibiting reductions of glutathione and NADP+ in the pentose phosphate pathway. (c) Its inhibition on the translation of GPx1, a major antioxidant enzyme in vascular cells, upregulates mitochondrial reactive oxygen species flux. (d) The enhanced expression of iNOS and uncoupling of NOS under make vascular inflammatory and generation of nitrogen reactive species. (2) Homocysteine encourages foam cell formation in atherosclerotic intima, which in turn promotes ROS production for LDL oxidation, SMC proliferation, and endothelial dysfunction. (3) Homocyteine accumulation impairs H2S (a vasodilator) production, which is also synergistic with AT1R, induced ET-1, attenuated NO/PGI2, etc., ensuring endothelial dysfunction, hypertension, and thrombosis. (4) Its thrombotic roles include (a) enhanced platelet activation, (b) enhanced coagulation as the result of increased TF expression, and/ or (c) attenuated fibrinolysis by (i) posttranslational modification of fibrinogen resistant to plasmin cleavage, (ii) increased TAFI activity, (iii) increased PAI-1 expression, and (iv) decreased tPA catalytic activity resulting from the homocysteine posttransloational modification of annexin A2, an EC coreceptor for tPA and plasminogen. (5) Homocyteine accumulation impairs angiogenesis known as angiostasis consistent with vascular dysfunction by (a) decreased glutathione peroxidase expression and consequent increased oxidant stress, leading to endothelial progenitor cell dysfunction and (b) decreased bioactive NO generation, increased expression and activity of MMP-2/MMP-9, and increased thrombospondin-1 expression. (6) Other related cardiovascular effects include intracellular Ca+2 mobilization, ER stress, activated AkT and p38 MAPK/ERK for VSMC proliferation, upregulated diverse pro-inflammatory signaling involving expression of proinflammatory gene (e.g., cytokines, VCAM-1, and CD40/CD40L) and transcriptional factors, plaque instability (anti-angiogenesis: increased MMP secretion & apoptosis), attenuated endothelial HO-1, suppressed PPARγ , etc. Some controversies however remain whether homocysteine lowering could really benefit to CVD [76].
Inflammation
Inflammation resulting from infection, tissue damages, hypercoagulability, platelet activation, etc. [77] produces an array of cytokines (e.g., TNF, IFNs, ILs, etc.), growth factors, and adhesion molecules (e.g., ICAM, VCAM, MCP-1, etc.). These cytokines decrease myocardial contractility, induce myocardial damage, and enhance the production of free radicals, which can also suppress myocardial function. Growth factor, for instance, TGFß enhances ET-1 and PAI-1 production, which in turn leads to endothelial dysfunction, hypertension, and thrombosis for atherosclerosis, hypertrophy, and HF. The upregulated signaling pathways (e.g., PI3K/AkT/mTORC1, MEK/ERK, JAK/STAT, etc. [77]) readily contribute to CVD pathogeneses (e.g., cardiac hypertrophy, AF, etc.). The lack of resolution of inflammation [77] also favors inflammatory responses leading to chronic diseases. For instance, atherosclerosis is a chronic inflammatory disease involving an array of inflammatory responses for its progression [78,79]. Oxidative stress-inflammation axis [23-26] further ensures CVD risk.
Obesity
Overweight (30> BMI >25) or obesity (BMI > 30) is associated with not only hyperlipidemia (e.g., hyperTG) but also other pathological states such as inflammation, oxidative stress, diabetes (diabesity), etc. Chronic mTORC1 activation in response to excessive nutrients is recognized as a pathogenic factor for obesity. In addition to genetic factors, overeating, and physical inactivity, gut microbiota play significant pathogenic roles. Obese microbiota (dysbiosis) with 50% reduction in abundance of Bacteroidetes and a proportional increase in Firmucutes bacteria [80] in addition to low in gut microbiota diversity. Obese microbiota, for instance, Lactobacillus and Mollicutes species within the Firmicutes family, significantly contribute to dietary energy harvesting from fermentation (SCFAs: butyrate and acetate production) of saccharide (e.g., fibers, pectins, etc.) or refined sugar (e.g., glucose, fructose, and sucrose), increased lipogenic enzymes (e.g., acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS), cholesterol synthesis, and transcription factor (ChREBP and SREBP1c) activation; all events favor energy production or body fat storage instead of expenditure not to mention about chronic low-grade systemic inflammatory state of obesity). Biochemically, obesity is often associated with decreases in the levels of skeletal muscle and abdominal tissue sirtuins (SirT), peroxisome proliferator-activated receptor coactivator (PGC-1a), mitofilin, transcription factor A mitochondrial (TFAM), uncoupling protein 1 (UCP1), and deiodinase [81]. Obesity with its frequent coexistence of sleep apnea, diabetes, hypertension, and coronary disease poses AF risk.
(1) Adipokines. Adipose tissue is a large endocrine system and adipocytes, especially white adipose tissues, produce a variety of biologically active adipokines (e.g., PAI-1, visfatin, resistin, leptin, and adiponectin) in addition to elevated TNF-a, IL-6/8, monocyte chemoattractant protein 1 (MCP-1), CRP, and osteopotin [82]. Expect adiponectin remaining low in obesity, most of which trigger inflammatory responses and CVD events. For instance, resistin promotes MFs up-taking OxLDL into foam cells, IL-6/IL-8/TNF-a production, angiogenesis (EC growth activation and migration), ET-1 release, and VSMC proliferation while potentiating CD40L effect. Visfatin promotes insulin resistance, endothelial dysfunction, VSMC proliferation, angiogenesis, and atherosclerotic plaque instability.
By contrast, adiponectin is considered protective, based on the potent insulin-sensitizing, antilipotoxic, anti-apoptotic, and antiinflammatory actions; it acts on different cell types and organs including muscle, heart, pancreas, liver, and adipocytes as key targets. Adiponectin attenuates TNF-a and IL-6 production while inducing expression of anti-inflammatory cytokines (e.g., IL-10 and IL-1 receptor antagonist). This anti-inflammatory adiponectin accounts for satiety, weight loss, anti-atherogenic, anti-diabetic, insulinsensitizing (attenuated insulin resistance), and hypolipidemic actions (lowering LDL-C and TG). Biochemically, adiponectin through AdipoR1 activates AMPK-PGC1a pathway or through AdipoR2 activates PPARa, exerting downregulated hepatic gluconeogenesis, antidiabetes, decreased insulin resistance and glucose intolerance. Increased FA oxidation is mediated by the increased expression of medium chain acyl-CoA dehydrognease (through AdipoR1 action), acyl-CoA oxidase, UPC-1 (through AdipoR2 action). The PGC1a activation by adiponectin essentially boosts mitochondrial proliferation and energy metabolism. Adiponectin also increases thyroid hormone synthesis, especially free thyroxine, and shares some physiological actions with thyroid hormones including body fat reduction, thermogenesis, and increased lipid oxidation [82]. Low adiponectin is associated with obesity, diabetes II, and insulin resistance.
(2) Increased oxidative stress. There is increased oxidative stress in obesity; increased ROS/RNS and their intermediates readily refuel chronic inflammation. Moreover, obesity per se drives M1 polarization that is characterized by iNOS and further produces TNFa, IL-1ß, IL-6, MCP-1, and O2•- [83].
(3) Inflammatory sequelae. Obesity is known as a low-grade chronic inflammatory disease [83-86], although inflammation is not proposed as an initial cause of obesity. Of particular importance, obesity triggers systemic inflammation in distant cells such as endothelium, arterial and bronchial smooth muscle, and pancreatic islets. Such systemic inflammation could lead to progression to diabetes (e.g., insulin resistance with activated inflammatory signaling pathways), atherosclerosis (endothelial dysfunction and hypertension). For instance, elevated fatty acid released from white adipocytes induces NLRP3 inflammasome activation and promotes proinflammatory cytokine IL1ß /18 secretion. Proinflammatory iNOS overexpression in obesity induces insulin resistance and offsets insulin actions, which is typically mediated by S-nitrosylation and inactivation of IRS1/2 and AkT as well as by many other canonical inflammatory mechanisms.
(4) Others. Its hypercoagulable (e.g., elevated FVII) and prothrombotic (e.g., elevated PAI-1) state also contributes to CVD risk. Increased ET-1 in obesity is readily involved in endothelial dysfunction for CVD development/progression.
Diabetes
Statistically, nearly 70% of diabetes population suffers from atherosclerosis, resulting largely from endothelial dysfunction, oxidative stress, and inflammation. Advanced glycation end-product (AGE; e.g., HbA1c) plays a central role in endothelial dysfunction and tissue damages, while low adiponectin associated with diabetes in part accounts for inflammation and reduced insulin sensitivity. Hyperglycemia, insulin resistance, and accompanied hypoglycemia impact on coagulation cascade, EC function, platelet and monocyte adhesiveness, macrophage function, and fibrinolysis, all leading and/ or contributing to CVD progression. For instance, EC activation results from elevated AGEs and cytokines. Shifting to aerobic glycoysis, enhanced pentose phosphate pathway, and polyol pathway of sorbitol and fructose production, diabetic ECs feature NAPDH consumption, glutathione depletion, and ROS accumulation [87]. In the presence of high glucose, eNOS undergoes O-linked glycosylation, becoming inactive and reducing NO synthesis. Thus, increased ROS and reduced NO favor diabetic hypertension, certainly contributing to CVD risk. In diabetes, modified fluid and cellular phases generate prothrombotic milieu, which also participates in atherogenesis. HF is a common CVD presentation in diabetes II whose duration exacerbates CVD risk/progression especially in women.
(1) Insulin resistance. Insulin resistance per se could trigger atherothrombosis [88]. (a) Impaired insulin sensitivity in VECs leads to increased FFA oxidation, ROS formation, and subsequent activation of detrimental biochemical pathways such as AGE synthesis, PKC activation, protein glycosylation as well as PGI2 downregulation, all of which blunt eNOS activity and lead to endothelial dysfunction. (b) Similar to obesity, excessive FFA availability triggers inflammation [89] and PAI-1 expression [61]. (c) Lack of insulin signaling in platelets impairs the IRS1/PI3K pathway, resulting in Ca2+ accumulation and increased platelet aggregation.
(2) Hyperglycemia. Diabetes often presents hyperglycemia [90] with elevated proinflammatory AGE that per se induce EC damages/ dysfunction, NOX (ROS production), iNOS, and cyclooxygenase (COX)-2. Hyperglycemia drastically impacts on global metabolism. High glucose exposure encourages sorbitol production continually metabolizing to fructose. In the polyol pathway, such conversion consumes NADPH, thereby increasing ROS accumulation; fructose promotes AGE formation for vascular damages. AGE signal through its receptor also activates inflammatory kinase (e.g., JNK or IκB kinase (IKK)) and in turn blocks IRS tyrosine phosphorylation, leading to insulin resistance.
Diabetic hyperglycemia activates CaMKII by O-linked glycosylation, thus posing risks for arrhythmia and HF. Hyperglycemia (e.g., HbA1c) also induces vascular damage. (a) Glucose-induced PKC activation upregulates ET-1 synthesis, favoring vasoconstriction and platelet aggregation. (b) PKC-dependent eNOS deregulation, ROS production by NOX, and p66Shc adaptor protein increase oxidative stress rapidly and inactivates NO leading to the formation of cytotoxic ONOO-. (c) PKC-mediated pro-inflammatory gene (e.g., MCP-1, VCAM-1, and ICAM-1) activation via NFkB signaling leads to monocyte adhesion and rolling responsible for initiating atherogenesis. (d) Impaired glucose-6-phosphate dehydrogenase (an antioxidant enzyme) in diabetes (hyperglycemia) facilitates oxidative status by inhibiting reductions of glutathione and NADP+ in the pentose phosphate pathway [87]. (e) In animal models, excess glucose encourages overproduction of mitochondrial ROS and phosphorylates VEGFR within the Golgi, leading to VEGFR degradation; VEGFR is essential for EC growth, survival, and function.
Diabetes per se readily poses thrombotic risk, which is mediated by the polyol pathway-induced ROS. Hyperglycemia augments vWF expression and secretion. Hyperglycemia increases glucose flux into the polyol pathway [87], in which aldose reductase and sorbitol dehydrogenase generate sorbitol and fructose in processes, using NADPH and NAD+ as cofactors. The process leads to an increase in oxidant stress; (a) the augmented formation of NADH leads to the production of ROS by NADH oxidase and (b) the depletion of NADPH causes reduction of cytosolic glutathione levels. Through the polyol pathway, hyperglycemia possibly via increased ROS induces c-Myc mRNA and c-Myc activation (phosphorylation) represses miR24 expression that in turn attenuates vWF expression and secretion from ECs. Thus, hyperglycemia-induced ROS elevates vWF plasma levels; vWF binding FVIII and platelets could contribute to diabetic hypercoagulability and thrombosis.
(3) Hypoglycemia. Hypoglycemia largely results from insulin therapeutics (e.g., insulin replacement, sulfonylureas, or glinides) or compromised glucose counterregulation upon absolutely damaged ßcells as in diabetes I and advanced diabetes II [91], which also leads to CVD risks [92]. (a) Hypoglycemic events could trigger inflammation by inducing the release of CRP, IL-6, and VEGF. (b) Hypoglycemia induces platelet and neutrophil activation. (c) The sympathoadrenal response during hypoglycemia increases adrenaline (e.g., epinephrine) secretion that could induce arrhythmias and increase cardiac workload. (d) The resulting endothelial dysfunction with suppressed vasodilation could certainly contribute to CVD risk.
(4) Others. Diabetes often associated with elevated homocysteine [74] triggers oxidative stress, endothelial dysfunction, and hyperhomocysteinemia consequence (see Major CVD risk under the section of Hyperhomocysteinemia) readily posing CVD threats in addition to prothrombotic (e.g., elevated PAI-1 and TxA2) and hypercoaguable [46] (e.g., elevated TAFI [93], TF expression, FVIIa, and vWF) state of diabetes.
Typical Pharmacological Approaches
Accordingly, cardioprotection aims at lowering CVD risks, blocking pathogenesis and progression as well as relieving symptoms. FDA has approved several drugs for combating hyperlipidemia. Anticoagulants, anti-platelet agents, and NSAID are widely recommended for thromboembolism. ACE inhibitors and ß-blockers are for hypertension. Antioxidants, NSAID, and steroids are for fibrosis. ACE inhibitors, ß-blockers, and antioxidants are for cardiomyocyte damage. ß-blockers are for arrhythmia, while ACE inhibitors are for conduction effects. For HF, ACE inhibitors, renine inhibitors, and angiotensin receptor blockers are recommended. Ntriuretic peptide ularitide, a chemically synthesized analogue of the naturally occurring vasodilator urodilatin, shows reductions in NTproBNP levels and reduces the long-term risk of cardiovascular death from acute HF. In addition, antioxidants could generally attenuate oxidative stress and inflammation. As expected, pharmacological agents often exhibit some degrees of adverse effects, if not all related to CVD risks.
Hypolipidemic effects
To aid hypolipidemic actions, FDA has approved several drugs for combating hyperlipidemia, which includes statins (1987), ezetimibe (2002), ApoB100 inhibitor (2013), ApoC3 inhibitors (2014), PCSK-9 inhibitors (2015), and many more to come. Others such as raising HDL and RCT (please refer to [94]) are beyond the scope of this review; for instance, ApoA1 mimetics (e.g., ETC-216, D4F from D-amino acids, etc.) could elevate HDL-C and reverse the progression of atherosclerosis in view of ApoA1 serving as a HDL progrogmmer for RCT by increased cholesterol efflux from MFs via ATP-binding cassette A1.
(1) Hypocholesterolemic actions. Concerning the lipid hypothesis, plasma cholesterol becomes a main target for lowering atherogenic risk. The strategies include the first generation of treatment with bile acid sequestrants (cholestyramine, colesevelam, colestipol, and colestimide), lowering cholesterol biosynthesis, and upregulation of LDL uptake. An antisense oligonucleotide that inhibits ApoB100 could also be employed for treatment of homozygous FH.
(a) Bile acid sequestrants (BAS). BAS directly binding bile acids in the intestine has been used for more than 50 years in treatment of hypercholesterolemia [95]. More than a simple resin, BAS exhibits diverse metabolic effects including energy expenditure in light of bile acids as integrated regulators of metabolism via induction of various signal transduction pathways. (i) The removal of bile acids activates hepatic CYP7A1, a regulatory enzyme for cholesterol catabolism to bile acid; mechanistically, decrease in bile acids transferred to the liver via the portal vein leads to such upregulation on the activity. As a result of facilitated cholesterol catabolism, LDL receptor expression is in turn upregulated via cholesterol homeostasis. (ii) The sequestrants also inhibits GI cholesterol absorption by preventing formation of micelles composed of bile acids in the intestinal lumen, which may also contribute to LDL-C lowering effect. (iii) Their hypolipidemic effects include decreases in the expression of SREBP-1c (a key lipogenic transcription factor), fatty acid synthase, SHP, and PEPCK (a key enzyme of gluconeogenesis) without significant effects on LXR and fatty acid oxidation (no effect on mitochondrial CPT-1). The side effects of BAS include flushing and diarrhea. Prolonged usage could lead to bleeding episode, which is proposed to be BAS binding to vitamin K, an essential cofactor for overall blood coagulant (FIX, FVII, FX, prothrombin, and protein C and S) function.
(b) Diverse statin effects. The current guidelines are set for initiating statin treatment [34]. (i) As a competitive inhibitor of HMG-CoA reductase, its classical cholesterol lowering action significantly inhibits hepatic de novo cholesterol biosynthesis, which dictates cholesterol homeostasis (in this case cholesterol acquisition) and in turn enhances LDL receptor expression (SREBP cleavage) for LDL-C clearance [96]. It also marginally increases HDL-C. In the context of its cholesterol lowering, statins eventually prevent inflammasome (NLRP3) activation from intracellular cholesterol accumulation, which in part unifies inflammatory atherogenesis. Stains indeed reduce CRP level. (ii) In addition to cholesterol content alterations on platelet membrane, statins through activation of the cAMP-eNOS/NO-cGMP pathway result in inhibition of the PLCγ2-PKC-p38MAPK-TxA2 cascade for its antagonisms against platelet activation and aggregation. (iii) Its anti-thrombotic actions include enhancement of the fibrinolytic system through induction of t-PA and downregulation of PAI-1 and TF expression. (iv) Its anti-hypertension is attributed to decreased ET-1 expression, upregulated eNOS and NO bioavailability, and downregulated AT1R expression. (v) Its reduction in vascular inflammation is mediated by inhibition of macrophage activation, proliferation, and MMP secretion. (vi) The inhibition of VSMC proliferation eases/arrests atherogenenesis. (vii) Statin promotes neovascularization through direct angiogenesis or mobilization of anti-inflammatory endothelial progenitor cells which bind and inhibit platelets for suppressed platelet activation/aggregation. (viii) Its antioxidant effect is mediated by NOX inhibition, HO-1 activation, and Nrf2 activation. The ability to promote S-nitrosylation of thioredoxin at Cys69 subsequently stimulates the antioxidative activity of thioredoxin to facilitate ROS scavenging. The antioxidant effects could account for their benefits to myocardial hypertrophy, fibrosis, and possibly HF. (ix) Statins induce cytoprotective genes, protect against vascular injury, and reverse endothelial dysfunction by inhibited RhoA, enhanced CD73 cell surface expression and activity (increased extracellular adenosine), improved vascular tone (increased NO biosynthesis), and inhibited platelet activation and leukocyte adhesion [97]. (x) Its broad anti-inflammatory actions include AMPK activation [98], eNOS activation, FOXO upregulation, IKK inhibition, IKK-independent NF B inactivation, JAK/STAT inhibition [99,100], PI3K/AkT/mTOR inhibition, increased IL-10, attenuated proinflammatory biomarkers (e.g., CRP, IL-6, and TNF) [100,101], suppressed CD40 expression [102], and depressed TF expression and its initiated blood coagulation. Statins also promote efferocytosis and cysteine S-nitrosylation of COX-2 for Rvs (e.g., 15-epi-LXA4) production [103], both of which are considerably anti-inflammatory. (xi) Immunologically, statin decreases MHC II expression, while it attenuates T cell activation by depleting membrane cholesterol and disrupting the integrity of lipid rafts that are essential to TCR signaling and costimulatory molecule assemblies [104]. Treg accumulation [105] by statin certainly presents anti-inflammation and immunosuppression.
However, its possible adverse effects could include liver injury (e.g. cholestatic hepatitis, hepatocellular injury, etc.), reversible muscle weakness, potential memory loss, increased food intake, damages on pancreatic cells, and significantly increased risks for newonset of diabetes [106] and autoimmune myopathy, which warrants further confirmation and investigation. It remains debatable whether long-term statins could pose the risk of diabetic retinopathy/catract as to diminished/attenuated downstream intermediate: lanosterol that could reverse/ resolve the catract. Precaution should also be taken with respect to statin uses in pregnancy as its potential risk of congential heart disease in offsprings. Scientific recommendations concerning statin interactions with diverse drugs in CVD patients have also been documented by American Heart Association [107].
(c) Enhanced LDL uptake. Alternatively by encouraging hepatic LDL uptake [108], recent clinical trials evaluate the efficacy of monoclonal antibodies against PCSK-9 in lowering plasma LDL-C. PCSK-9 binds EGF-A domain of hepatic LDL receptor to cause LDL receptor internalization, blocked recycling, and degradation by Idol, which virtually dampens LDL-C clearance from circulation. PCSK-9 inhibition includes anti-PCSK9 mAb (e.g., evolocumab, alirocumab, and bococizumab), PCSK-9 antisense oligonucleotides, PCSK-9 RNA interference (inclisiran), or other developing molecular manipulations. Consistently, loss-of-function PCSK-9 mutation (e.g., R46L) in ~2% of black population has shown substantially lowered cholesterol offering lifelong cardioprotection [109]. In the presence of a statin, PCSK-9 inhibitors or antibodies could even maximize plasma cholesterol lowering effect. Extending beyond lipid homeostasis, PCSK-9 is implicated as a regulator of glucose metabolism, liver regeneration, and susceptibility to HCV infection.
Interestingly, evinacumab, a fully human ANGPTL3-blocking antibody, effectively lowers not only TG but also cholesterol. Accordingly by lowering LDL-C, ApoB levels, non-HDL-C, HDL-C, and TG, ANGPTL3 inhibition exhibits additional benefits to homozygous FH already receiving statins, ezetimibe, lomitapide, PCSK9 monoclonal antibodies, or a portacaval shunt.
(d) Downregulation on dietary cholesterol absorption. Niemann-Pick C1-like protein 1 serves as a saturable transporter on microvilli to control dietary cholesterol inputs. The inhibitor (e.g., ezetimibe) could lower LDL-C by nearly 18% in addition to statin monotherapy. In view of plant sterol absorption sharing this transportor, dietary phytosterol such as ß-sitosterol could competitively reduce zoosterol absorption to some degree, if not all.
(2)Lowering plasma TG. Unlike cholesterol biosynthesis, there is no single regulatory enzyme in TG biosynthetic pathway, which can be targeted for lowering plasma TG supply. Combating hyperTG could be achieved by ApoC2 and ApoA5 activators and ApoC1/3 inhibition as well as ANGPTL3/4 blockade, all of which upregulate LPL activity. (a) ApoC3 inhibitor (antisense oligonucleotide: ISIS 304801 directed against ApoC3) shows 60-70% decrease in ApoC3, reducing CVD risk. (b) ANGPTL3 is a negative regulator of LPL and its loss-of-function consistently reduces TG and shows small reduction on LDL-C and HDL-C; ANGPTL3 inhibition thus could lead to benefits (e.g., reduced hyperTG/homozygous FH) to CVD. Anti-ANGPTL3 mAB (evinacumab; REGN1500) targeting plasma ANGPTL3 shows (i) TG reduction mediated at least in part through disinhibition of LPL and subsequent hydrolysis of TG-rich lipoproteins, (ii) HDL-C reduction mediated by disinhibition of endothelial lipase, and (iii) reduction in LDL-C by unknown mechanisms with additional benefits to FH already receiving statins, ezetimibe, lomitapide, PCSK9 monoclonal antibodies, or a portacaval shunt. Anti-ANGPTL3 mAbs, however, have no effects on hepatic TG content. (c) ANGPTL3 antisense oligonucleotides target hepatic ANGPTL3 mRNA, thus inhibiting hepatic ANGPTL3 expression. Such hepatic inhibition of ANGPTL3 leads to significant decreases in plasma levels of TG, LDL-C, non- HDL-C, VLDL-C, total ApoB, and ApoC3, which accompanies by decreased hepatic VLDL-TG secretion, decreased liver TG content, increases in insulin sensitivity, as well as increased plasma LPL activity for reduction in atherosclerosis progression. Anti-NAGPTL4 mAb (REGN1001) is developed and tested for such application of lowering TG in mice and monkeys. (d) Lomitapide/ implitapide (microsomal triglyceride transfer protein (MTP) inhibitor; BMS 212122) reduces ApoB-containing lipoprotein; MTP expressed in the liver, intestine and the heart transfers TG, PLs and cholesteryl esters to the ApoB in endoplasmic reticulum for the proper assembly of CM and VLDL, both of which are precursors for LDL. CM, VLDL, and LDL are TGrich and source of plasma TG. In addition, fibrate derivatives (e.g., bezafibrate) are of classical TG-lowering agents.
(3)Attenuating LDL. (a) ApoB is a programmer of LDL metabolism; inhibition on ApoB production is for treating homozygous FH. By targeting ApoB synthesis/secretion, antisense oligonucleotide (mipomersen) lowers LDL production and increases LDL fractional catabolic rates. Such targeting ApoB synthesis may lower levels of ApoB lipoproteins without necessarily reducing VLDL secretion, thereby lowering the risk of steatosis. (b) MTP inhibitor (implitapide; BMS 212122) reduces ApoB-containing lipoproteins (CM, VLDL, and LDL) for treating homozygous FH and athosclerosis regression.
(4)Reducing Lp [a]. ISIS-Apo (a) Rx, an antisense oligonucleotide targeting Apo[a] designed to reduce liver synthesis of Lp[a], shows benefits in clinical trials by decreasing plasma Lp[a] concentration by 40 to 80%. Interestingly, PCSK-9 inhibitors reduce Lp[a] by nearly 30 %, which is proposed to be mediated by Lp[a] internalization/ catabolism via LDL receptor.
Anti-thrombosis approach
Anti-thrombotic therapy/prophylaxis is recommended for reducing MI, ACS, and PCI. In general, aspirin is included in antithrombotic plan, while anticoagulants (e.g., anti-FXa, heparin, low molecular weight heparin (LMWH), and warfarin) are suggested for deep vein thrombosis. Warfarin and anti-FXa are also currently in the practice for AF. FXa and thrombin inhibitors are secondary prophylaxis against venous thromboembolism, while anti-platelet agents are recommended for atherothrombosis under high shear stress; they, however, could show similar efficacy under either condition according to recent clinical studies. The utility of thrombolytic agents (e.g., streptokinase, urokinase, and TPA) for lysing fibrin clots is also recommended.
(1) Anticoagulation. Typical oral anticoagulants include dabigatran, rivaroxaban, apixaban, and warfarin, while injectable anticoagulants include unfractionated heparin or low-molecularweight heparin (LMWH), bivalirudin (direct anti-thrombin inhibitor), and pentasaccharide fondaparinux (anti-FXa) [110]. They typically treat MI, acute coronary syndromes, during or after PCI.
Among which, warfarin is commonly used for thrombotic prophylaxis, AF, etc. Warfarin and its analogs block the K cycle by targeting and inhibiting vitamin K epoxide reductase, thereby showing anti-thrombolic effect of reduction in overall coagulant (FIX, FVII, FX, prothrombin, and protein C and S) function. Thus, over- or under- dosing could relate to bleeding or thrombosis, respectively [111]. The reductase mutation could lead to warfarin resistance, while gain-of-function mutation of CYP2C9 (CYP2C9-1) could effectively degrade warfarin. In addition to its pharmacogenetic concerns, warfarin per se binds to albumin, and only about 3% is in free and pharmacologically active form. A number of medications can displace warfarin, leading to its increased activity and subsequent increased rate of degradation. Diet, specifically the intake of foods containing vitamin K, can offset the effect of the daily dose of the vitamin K antagonist. Age, weight, and sex are also factors that could influence the dosing, while warfarin resistance results from pharmacogenetic variations on the epoxide reductase. On the other hand, prolonged and high dose exposure could show bleeding episode; INR monitoring is required (INR < 2 presents thrombosis, while INR ≥ 4 indicates bleeding).
Recent clinical trials show that FIX antisense oligomer doesn’t cause bleeding. Anticoagulant heparin often induces thrombocytopenia that is prothrombotic associated with antibody (IgG2 FcγRIIA 131R) against heparin-PF4 complex triggering platelet aggregration. For bleeding episodes, antidotes idarucizumab and andexanet alfa (andexanet) could be required for anticoagulants dabigatran and FXa inhibitors, respectively.
(2) Anti-platelet. Typical antiplatelet agents include aspirin, clopidogrel, prasugrel, dipyridamol, abciximab, cilostazol, and ticagrelor. Inhibition of thrombin by anticoagulants results in less platelet activation, although unfractionated heparin could activate platelets (e.g., PF4). Anticoagulants: unfractionated heparin, LMWH, and direct thrombin inhibitors (lepirudin, argatroban, bivalirudin, and dabigatran), however, are not specific antiplatelet drugs. For atherothrombosis, (a) antiplatelet therapy by ADP receptor inhibitors (ticagrelor, prasugrel, clopidogrel, or cangrelor) mediates their actions by inhibiting guanine nucleotide-binding protein-coupled purinergic receptor P2Y12 through an irreversible binding. These ADP receptor inhibitors also suppress CD39 that converts extracellular ATP to ADP, thus limiting ADP supply. Similarly, reversible P2Y12 antagonism by ticagrelor (cyclopentyl-triazolo-pyrimidine) or cangrelor (ATP analog) could be used. (b) Glycoprotein IIb/IIIa inhibitors/ antagonists (e.g., abciximab, eptifibatide, tirofiban) essentially block the binding of FBG and, at high shear, vWF to GPIIb-IIIa on adjacent platelets and thereby block platelet-to-platelet aggregation, specifically inhibiting platelet reactivity [112]. Abciximab is the Fab fragment of the chimeric human-murine monoclonal antibody 7E3, whereas eptifibatide is a KGD-containing cyclic hexapeptide, smallmolecule inhibitor. Abciximab and eptifibatide bind to the GPIIb- IIIa (integrin aIIbß3) receptor of human platelets, inhibiting the final common pathway of platelet aggregation in response to all agonists (e.g., ADP, collagen, thrombin, or fibrin). Tirofiban is a nonpeptide mimetic tyrosine derivative based on RGD, which selectively binds to GpIIb-IIIa. Interestingly, in vitro prasugrel metabolite (R-138727) in combination with abciximab or eptifibatide shows anticoagulant potentials of suppressed platelet TF and membrane surface phosphatidylserine expression and reduced thrombin generation [113]. (c) Dipyridamole is a pyrimidopyrimidine derivative with vasodilator and antiplatelet properties, which is proposed to inhibit PDE and accummulate cAMP (a vasodilator) or direct stimulate PGI2 synthesis and reduce PGI2 degradation. Similarly, cilostazol also exhibits antiplatlet and vasodilatory properties. (d) Bloodthinner aspirin (lower dose) as an anti-platelet agent effectively blocks platelet COX-1 for TxA2 formation. Mechanistically, low-dose aspirin acetylates serine 529 on COX1 and inhibits COX1, thereby blocking TxA2 generation; Optimal/effective aspirin doses have been recommended by the American College of Chest Physicians (2012) for atherothrombosis (MI, stroke, or vascular death), AF, and pulmonary embolism preventions. It has been reported by clinical evidence that aspirin plus P2Y12 antagonist therapy with a GPIIb-IIIa antagonist greatly enhances platelet inhibition [114]. (e) Caplacizumab, an anti-vWF humanized single-variable-domain immunoglobulin, targets the A1 domain of vWF, preventing interaction with the platelet glycoprotein Ib-IX-V receptor and platelet aggregation. (f) Vorapaxar (tricyclic himbacine derivative) blocks platelet PAR1, a known thrombin receptor for platelet activation. (g) Other such as NO donors could inhibit platelet activation/aggregation/secretion via classical NO/cGMP/PKG pathway.
(3) Thrombolysis. tPA (e.g., streptokinase, urokinase, alteplase, reteplase, tenecteplase, anistreplase, desmoteplase, or viprinex) promoting plasmin production from plasminogen is used mainly for stroke patients, but also for thrombolytic therapy [115]. It has been reported that exogenous tPA could cross both intact and the damaged blood brain barrier to reach the brain parenchyma, influencing brain functions and dysfunctions [116]. Molecular mechanisms underlying such neurotoxicity within the neurovascular unit however have not been fully understood. APC could offer some benefits over tPA, while anti-vWF therapy (caplacizumab) could complement t-PA in thrombolysis.
PAI-1 and TAFI also become targets for thrombolytic therapy. Anti-diabetes agents: thiazolidinediones (e.g., troglitazone or pioglitazone) could reduce PAI-1 levels. Ramipril for acute MI lowers PAI-1 level. Statins (e.g., atorvastatin and rosuvastatin) as well as fibrates decrease PAI-1. Bis-arylsulfonamide and derivatives as well as indole oxoacetic acid derivative (tiplaxtinin) have been shown as PAI-1 inhibitor in vivo and in vitro [117]. Oral TM5007 [118] inhibits PAI-1 activity and increases fibrinolysis in nonhuman primate models by interaction with the strand 4 position of the A ß-sheet of PAI-1. Non-pharmacological approach includes physical exercise which is known to enhance fibrinolytic activity by increasing t-PA activity and lowering PAI-1.
TAFI inhibition could facilitate tPA-induced fibrinolysis; many TAFI inhibitors [119] (e.g., guanidinoethyl-mercaptosuccinic acid, e-amino caproic acid, potato tuber carboxypeptidase inhibitor, DL- 2-mercapto methyl-3-guanidinoethyl-thiopropanoic acid, leech carboxypeptidase inhibitor, tick carboxypeptidase inhibitor, SAR- 104772, compound-8/14, UK-396,082, AZD-9684, BX 528, and EF6265) await confirmation for their clinical applications.
Anti-hypertension approach
In general, monotherapy could ease hypertension in approx. onethird hypertensive population. Another one-third patients could need two drugs, and the rest might require as many as at least three agents. Combination therapy includes renin inhibitor/calcium-channel blocker, AT1R antagonist/diuretic, AT1R antagonist/calciumchannel blocker, AT1R antagonist/calcium-channel blocker/diuretic, or calcium-channel blocker/renin inhibitor/diuretic.
The conventional treatments targeting RAAS include diuretics (e.g., hydrochloro-thiazide), ß-blockers, ACE inhibitors, AT-II inhibitors, rennin inhibitors, AT-II receptor blockers, Ca+2 channel blockers (e.g., amlodipine), a-blockers, and a/ß -blockers. Diuretic agents and ACE inhibitors as well as ß-blockers remain mainstay for anti-hypertension. Novel ones in clinical development include AT1R blocker, AT2R agonist (e.g., compound 21), aldosterone antagonists, aldosterone synthase inhibitor, natriuretic peptide agonist, soluble epoxide hydrolase inhibitor, renin inhibitors, AT II vaccination, NO induction, and many novel strategies. Novel antihypertensive sets with dual activity include AT-receptor blocker/neutral endopeptidase inhibitor, AT-receptor blocker/ETA receptor blocker, and ETconverting enzyme/neutral endopeptidase inhibitor [120].
Apart from NO donors being hypotensive, sGC activator (e.g., cinaciguat) and stimulator (e.g., riociguat) readily elevating cGMP are currently in human trials. PDE5 inhibitors (sildenafil and vardenafil) are cGMP structural analogues, competitively binding to the catalytic site and inhibiting PDE5 to result in relaxation. In view of cAMP hypotensive effect, PGI2 analogues (epoprostenol or iloprost) elevating cAMP levels are of clinical benefit for anti-hypertension. Andrenomedullin, a 52 amino-acid peptide and a member of the calcitonin gene-related peptide superfamily, exhibits vasodilation in addition to its stimulation on NO/cGMP and mediation by PI3K/AkT pathway. Forskolin, an adenylate cyclase activator, stimulates cAMP formation. Alternatively, inhibition of PDE3/4 by rolipram leads to cAMP accumulation, which also contributes to EC-independent SMC relaxation.
Antagonisms against diverse CVD types
(1) Amiodarone and dronedarone are effective, while -blockers, digoxin, ACE inhibitors, and AT receptor blockers are classical treatments for anti-arrhythmia. ACE inhibitors reduce fibrosis. Oral anticoagulants (warfarin, direct thrombin inhibitors: ximelagatran), aspirin, antiplatelet (e.g., clopidogrel) show a risk reduction of arrhythmia. (2) For arrhythmia commonly associated AF, novel oral anticoagulants (warfarin or anti-FXa: rivaroxaban) are appropriate for “nonvalvular” AF, while warfarin remains effective for “valvular” AF. It is noted that direct-acting oral anticoagulants (apixaban, dabigatran, or rivaroxaban) for AF could lead to poor renal function; dose reduction/readjustment is needed for achieving non-bleeding without a renal dysfunction. Others such as dofetilide, dronedarone, flecainide, propafenone, sotalol, or amiodarone maintain sinus rhythm in patients with AF. However, aspirin should no longer be considered for stroke prevention in AF patients. For nonvalvular AF, long-term antithrombotic therapy is recommended including oral anticoagulation with warfarin, dabigatran, rivaroxaban, or apixaban and/or antiplatelet therapy with clopidogrel. (3) For combating hypertrophy, nifedipine inhibits L-type Ca+2 channel and calcineurin/ NFAT-mediated hypertrophic gene expression. ACE inhibitors also ease AT-II-mediated hypertrophy. Anti-hypertensive ACE inhibitors and anti-thrombotic dipyridamole could be included, while sildenafil enhances not only NO bioavailability but also VEGF signaling for angiogenesis, contributing to the regression of pathological hypertrophy [121]. (4) For angina treatment and attenuation, nitrates, calcium-channel blockers, and -blockers ranolazine, nicorandil, ivabradine reducing heart rate and trimetazidine are recommended as first line therpy with ranolazine as second line. (5) HF therapy could include -blockers, AT-receptor blockers (interfering with the action of AT-II at its type 1 receptor), ACE inhibitors, loop diuretic, spironolactone (mineralo-corticoid-receptor antagonist), nesiritide (a recombinant B-type natriuretic peptide as a vasodilator), neprilysin (neutral endopeptidase) inhibition with LCZ696. In addition, three types of cardiac devices are also available, which includes the left ventricular assist device, the implantable cardioverter-defibrillator, and cardiac resynchronization therapy by biventricular pacemakers.
Anti-inflammatory approach
Glucocorticoids remain the most effective therapy widely used in anti-inflammatory practices [122]. (1) Genomically, it inhibits the gene expression of pro-inflammatory factors (IL-1, IL-2, TNF-a, IFN-γ, prostaglandins, NOS, adhesion molecules, etc). Glucocorticoids inhibit the production of TSLP, cytokines, chemokines, adhesion molecules, and other inflammatory mediators, which is proposed to mainly repress key proinflammatory transcription factors: NFkB and AP-1. For instance, they suppress NFκB-dependent transcription by upregulating MAPK phosphatase-1 to dephosphorylate p38 MAPK. As a result of downregulated NFkB, glucocorticoids also suppress COX-2, iNOS, and ICAM-1 expression. Similarly, corticosteroids induce MAPK phosphatase 1, inhibit JNK, inactivate NF B and AP-1, block PLA2, COX-2, and lipocortin-1, and reduce PG and LT biosyntheses. As the result of suppressed production of IL-1, TNF-a, GM-CSF, IL-3, IL-4, IL-5, and CXCL 8, corticosteroids readily exhibit antiinflammation. Furthermore, 11ß-hydroxysteroid dehydrogenase type 1 converts glucocorticoids to cortisone that modulates intracellular access of glucocorticoid receptors, amplifying glucocorticoid effects [123]. (2) Its non-genomic post-transcriptional effects include (a) blocking AA release/generation by cPLA2 inactivation that results from inhibition of the phosphorylation of MEK1 and suppression of the MEK substrate ERK1, (b) inhibition of COX2 expression for suppressed PGE2 production, (c) shifting AA metabolism toward anti-inflammatory endocannabinoid synthesis by activating PI-PLC hydrolysis and promoting 2-arachidonoyl-glycerol production while upregulating PKA for anandamide production, and (d) promoting the resolution of inflammation by inhibiting the vasodilation and increasing vascular permeability following inflammatory insult while decreasing leukocyte emigration into inflamed sites. (3) From immunological viewpoints, glucocorticoids suppress T effectors; T effector proliferation requires IL-2, IL-4, IL- 5, IL-17, and IFNγ. Glucocorticoids activate MF phagocytosis of apoptotic cells while increasing the expression of IL-1 decoy receptor and promoting MF to release anti-inflammatory IL-10 and TGFß. However, glucocorticoids are known to widely affect metabolisms; in fact, glucocorticoids could associate with high BP, blood glucose, and TG levels, low HDL-C levels, and many forms of lipodystrophy that are associated with metabolic disorders and premature atherosclerosis. Glucocorticoids also induce insulin resistance by affecting insulin signaling components (IRS-1, PI3K, PKB, GSK- 3, FYN, AMPK, p70S6k, increased JNK phosphorylation, etc.) in addition to its induction on osteoporosis.
Apart from classical COX inhibition for attenuating inflammatory PGs and LTs species, aspirin (non-steroid antiinflammatory drug) effects include AMPK activation, suppressed TNF secretion, and the serine acetylation of COX-2 for the formation of antiinflammatory resolvin (Rv) and 15-epi-LXA4 [77,124]. An anti-inflammatory clinical trial on canakinumab, a humanized monoclonal antibody against IL-1, has evaluated for its application to atherothrombosis. Canakinumab significantly attenuates CRP (<2 mg/L) and atherosclerosis, which underlines the pathogenic role of inflammation in atherosclerosis [78]. Canakinumab once every 3 months also broadly reduces the rate of a composite of nonfatal MI, nonfatal stroke, or CVD death.
Anti-diabetes approach
Incretin therapies (GLP-1 analogues (liraglutide), GLP-1R agonists, exenatide, dipeptidyl peptidase 4 (DPP-4) inhibitors, amylin analog (e.g., pramlintide)), biguanides (e.g., metformin), a-glucosidase inhibitors, bile acid sequestrants (e.g., colesevelam), and thiazolidinediones are considered low-risk for hypoglycemia, while insulin replacement and sufonylurea (glipizide) may have episodes of hypoglycemia. Among approx. 12 classes of drugs in diabetes II management, metformin remains mainstay. (1) Metformin represses the expression of gluconeogenic enzyme glucose-6-phosphatase, which is mediated by AMPK activation phosphorylating HDAC and CREB-regulated coactivator/TORC2 complex for blocking FOXO transcriptional activity (nuclear acetylated FOXO) and suppressing PGC-1a expression, respectively. In addition, metformin alters gut microbiota landscape toward healthy GI environment for GLP-1 and peptide tyrosine-tyrosine secretion from enteroendocrine L-cells while inhibiting DPP-4, an enzyme degrading GLP-1. It, however, could be associated with weight gain. (2) PPARγ ligands (e.g., thiazolidinedione: rosiglitazone and pioglitazone) are therapeutical targets for antidiabetes; PPARγ is able to increase insulin sensitivity by decreased insulin resistance-inducing adipokines (TNF, IL-1, and resistin) and increased adiponectin (insulin sensitizing). Its activation by PPARγ ligands, however, suffers adversal effects of weight gain, fluid retention, and CVD risk. (3) Long-acting GLP-1 mimetics (liraglutide) exhibits anti-diabetes in view of the abilities to stimulate insulin secretion, lower postprandial glucagon levels, slow gastric emptying, and reduce appetite. Liraglutide can also be used for effectively and safely treating nonalcoholic steatohepatitis. Liraglutide, however, might be associated with increased cancer risk, awaiting further confirmation. (4) DPP-4 inhibitors (saxagliptin or alogliptin) sustaining GLP-1 bioavailability could increase risk for congestive HF. (5) Dapagliflozin inhibits sodium-coupled glucose transporter type 2 in the renal proximal tubule, preventing the reabsorption of glucose into blood. However, its side effect includes increased glucagon secretion from cells, which may offset its efficicay.
Anti-obesity approach
Apart from natural antagonisms (e.g., exercise, calorie restriction/ reduction, dietary approaches (e.g., nutrient supplementation with (-) hydroxycitric acid, minerals, prebiotics, probiotics, etc.)) against obesity, several pharmacological agents are available. For instance, orlistat (xenical) inhibits pancreatic lipase, while serotogenic drugs: lorcaserin (Belviq), amphetamines, sibutramine, and others suppress appetite. CCK mimetics could supress food intake. Thermogenic drugs favoring fat burning are also considered anti-obesity. GLP-1 mimetic liraglutide is currently in clinical trials for its weight loss management; liraglutide could also be expected as anti-diabetes in view of abilities of GLP-1 mimetics to stimulate insulin secretion, lower postprandial glucagon levels, slow gastric emptying, and reduce appetite. Liraglutide, however, might be associated with increased cancer risk, awaiting further confirmation.
Key Biological Actions of Polyphenols
Figure 1 (right panel) summarizes diverse effects of polyphenols including classical anti-oxidation and AMPK- dependent and independent actions that are categorized into relevant biological functions in relation to cardioprotection.
Biology of polyphenols
Phytochemical polyphenols are plant secondary metabolites for colors and/or for defense against stressors such as pathogens. Phenolic compounds generally assume the responsibility for most fruit browning; polyphenol oxidase oxidizes polyphenol into quinones, highly reactive compounds that polymerize spontaneously to form brown pigments.
The complex polyphenol superfamily typically includes three categories: flavonoids, non-flavonoids, and phenolic acids. (1) Flavonoids can be sub-classified into catechins (found in green and white tea, grapes, cocoa, lentils, berries), iso/flavanones (naringenin, hesperetin; found in oranges, grapefruit, lemon, etc.), flavanols (kaempferol, quercetin, myricetin, isorhamnetin; found in green vegetables, apples, berries, onions, chocolates, tea, red wine, etc.), and anthocyanins (found in berries, red grapes, red wine, etc.). Catechins further comprise catechin, epicatechin, gallocatechin (GC), and epigallocatechin (EGC) and their gallates (EGCG). Proanthocyanidins are traditionally considered to be condensed tannins. (2) Non-flavonoids contain three sub-groups: resveratrol (found in grape skin, red wine, nuts), curcumin (found in turmeric plants, mustard), and coumarin (found in licorice, strawberries, apricots, cherries, cinnamon). (3) In the category of phenolic acids, (a) ellagic acid is found in walnuts, strawberries, pomegranates, cranberries, blackberries, guava, or grapes, and (b) tannic acid is found in nettles, tea, or berries. (c) Gallic acid is found in tea, mango, strawberries, rhubarb, and soy, while (d) caffeic acid can be found in blueberries, kiwis, plums, cherries, and apples.
It remains largely unclear whether lipophilic polyphenol absorption could share GI fat absorption through intestinal lymphatic pathway [125,126]. For instance, it is estimated in human that its serum concentration is near 70% of ingested resveratrol through the extrahepatic cycle delivery by lipoprotein. It is also proposed that fermented fruit juices often supply more polyphenols.
Polyphenol receptor
Limited research remaining elusive is available concerning polyphenol receptor(s); thus far, 67-kDa laminin receptor functions as a cell-surface EGCG receptor and EGCG is able to activate this laminin receptor signaling [127,128]. It is also possible that polyphenol affects a wide range of cell functions through simple diffusion or membrane lipid raft.
Classical antioxidation
The structural signatures of polyphenols afford radical scavenging as well as metal chelating as either primary or secondary antioxidants, arresting free radical chain reactions in biological damages. They are also able to inhibit ROS production from mitochondrial respiration, respiratory burst, and xanthine oxidase. Their antioxidant potentials are further enhanced by upregualtions on endogenous antioxidant enzymes responsible for ROS detoxification. For
instance, resveratrol downregulates NOX2 and NOX4 as a result of upregulating superoxide dismutase (SOD), glutathione peroxidase 1 (GPx1), and catalase. The classical antioxidant actions of polyphenols certainly complement their anti-inflammatory efforts by disrupting ROS-inflammation cycle [23-26].
(1) radical-scavenging. The structural features of poly hydroxyl groups on aromatic (phenyl) ring(s) make polyphenolic compounds much easier undertaking oxidation, exhibiting radical-scavenging of OH•- and NO•-. Some hydroxyl(s) depending on the adjacent chemical groups (e.g., methoxy) or positions (e.g. ortho) are even more potent for radical-scavenging activity. For instance, the orthomethoxy group in curcumin can form an intramolecular hydrogen bond with the phenolic hydrogen, making the H-atom abstraction from the orthomethoxyphenols. The H abstraction from these groups is responsible for the remarkable antioxidant activity. The trihydroxyl group on the B ring and the gallate moiety esterified at the 3’ position in the C ring of EGCG are believed to contribute to its scavenging activity.
(2) Metal chelating. Polyphenols quench the Fenton reaction to attenuate oxidative stress. In the classical Fenton reactions, transition metal: Fe+2, Cu2+, Co2+, Ti3+, Cr5+, or V2+ readily drives OH•- formation from H2O2 [129, 130]. Curcumin binds and chelates transition metal (Cu2+ and Fe2+) ions. Similarly, EGCG chelates Fe2+; EGCG is the most potent inhibitor of Fe2+-induced DNA break among polyphenols.
(3) NOX inhibition. Resveratrol [131,132], curcumin [131], apocynin [133], and many other polyphenols are able to inhibit NOX. For instance, curcumin [132] decreases NOX subunit (e.g., p67phox, p22phox, and gp91phox) expression, while resveratrol suppresses p47phox expression, both of which attenuate the generation of O2•- during innate responses to infection.
(4) Attenuation on mitochondrial respiration. By blocking the respiratory chain and ATPAse at the inner mitochondrial membrane, polyphenols thus inhibit mitochondrial ATP synthesis, attenuating mitochondrial ROS production such as O2•- and H2O2.
(5) Inhibition on xanthine oxidase. Polyphenols (e.g., resveratrol analogs [134], curcumin [135], EGCG [136], phenolic acids [137], capsaicin [138], quercetins [139], anthocyanins [139],) all inhibit xanthine oxidase, a ROS producing enzyme.
(6) Upregulations on endogenous antioxidant enzymes. In vivo, ROS detoxification by curcumin may be mediated through antioxidant enzymes such as superoxide dismutase (SOD), catalase, glutathione (GSH)-peroxidase (Px), and HO-1, which is believed to be mediated by nuclear factor erythroid 2-related factor 2 (Nrf2) and FOXO activations and other target antioxidant gene expression [140]. As a Michael acceptor, curcumin reacts with GSH (γ-Lglutamyl- L-cysteinylglycine, the main non-protein thiol found in cells) and thioredoxin. Similarly, resveratrol stimulates endogenous antioxidant enzymes (e.g., MnSOD and catalase) and antioxidant gene (e.g., NQO-1 and GST-P1) expression, while EGCG increases SOD and GSH-Px activities with increased cellular GSH.
Broad effects on cell signaling: Polyphenols generally inhibit mitochondrial ATP synthesis to trigger AMPK activation, involving divergent downstream signaling cascades such as SirT1 activation, eNOS induction, anti-inflammatory relevance (e.g., NFkB, COX, and iNOS inhibitions), FOXO upregulation, promoting p53 pathway, LPL upregulation and suppressed ANGPTL4 mRNA expression, CREBP activation, mTORC1 inhibition, SREBP-1c inactivation, PPARγ inactivation, HIF-1a repression, adiponectin elevation, and autophagy activation.
In addition, a wide-range of AMPK-independent actions [141] include PI3K/AkT inhibition, direct mTORC1 inhibition,ß-catenin inactivation, FOXO activation, PDE inhibition, ACE inhibition, attenuated ET-1 expression, proapoptosis, JNK or IKK inhibition, Nrf2 activation, Janus kinase (JAK)/signal transducer and activator of transcription (STAT) inactivation, MAPK inactivation, suppressions on TLR expression and high mobility group box-1 (HMGB1) release, modulation on BK+ channels as well as many non-signaling related other biological effects including secretase inhibition, monoamine oxidase inhibition, inhibition on a-glucosidase, anticoagulation, antithrombosis, epigenetic modulation, and gut microbiota alteration.
(1) AMPK activation. The majority of polyphenols inhibit mitochondrial ATP synthesis by blocking the respiratory chain and ATPAse at the inner mitochondrial membrane, thus activating AMPK. Additionally, resveratrol [142] inhibits cAMP-degrading PDEs (e.g., PDE4), resulting in accumulated cAMP that activates Epac1 for in turn stimulating PLC. The resulting phosphorylated ryanoidin receptor 2 triggers Ca2+ channel releasing CaM+2 from ER. The activated CamKKß then phosphorylates and activates AMPK. AMPK activation leads to SirT1 and FOXO activation as well as mTOR inhibition. AMPK activation could shift MF M1 to M2 polarization. Anti-inflammatory M2 MFs (as well as Treg) feature Th2 responses, resolving Th1 inflammation, immune tolerance, and profibrotic actions (tissue repair and remodeling) with high efferocytosis.
(2) SirT1 activation. Resveratrol, an allosteric activator of SirT1, increases the catalytic activity of SirT1. SirT1 substrates (e.g., PGC-1a and FOXO3a) facilitate resveratrol-mediated SirT1 activation. Resveratrol triggers an array of signal cascade to exhibit metabolic benefits via elevated NAD+ and enhanced SirT1 activity, known as AMPK-SirT1-PPARγ coactivator 1a (PGC-1a) axis. While phosporylating PGC-1 AMPK increases NAD+ levels and activates SirT1 that deacetylates PGC-1a. It is also proposed that polyphenol could inhibit poly(ADP-ribose) polymerase-1, thereby increasing NAD+ level, a cofactor for SirT1 activation.
SirT1 activation [142,143] triggers diverse signaling including AMPK activation, NFkB inactivation, eNOS activation, p53 activation, tumor suppressor FOXO upregulation for antioxidant enzyme (MnSOD) expression, early Treg differentiation (antiinflammation), suppressed lipogenesis (PPARγ inactivation) [144], longevity, etc. Thus, metabolic benefits such as anti-aging, anti-diabetic, and increases in FA oxidation, gluconeogenesis, and mitochondrial biogenesis and functions could result from indirectly activated SirT1 via competitive inhibition of cAMP-phosphodiesterases (PDE4) by polyphenol [142,143]. In these regards, resverstrol also mimics caloric restriction (e.g., short-term fasting or exercise).
(3) NFκB inactivation. NFκB inactivation by polyphenols is mainly mediated by PI3K/AkT inhibition, FOXO activation, and IKK inhibition. Curcumin, for instance, inhibits TLR4-induced IKK / phosphorylation; the resulting blocked release of IκB results in NFκB inactivation. Alternatively, curcumin inhibits AkT and its consequent I B phosphorylation, similarly leading to NFκB inactivation [145-148].
(4) PI3K/AkT inhibition. Polyphenols generally inhibit PI3K/ AkT signaling cascade; its downstream effects include IKK inhibition and FOXO activation, exhibiting antiinflammation. Synergistically with AMPK activation, PI3K/AkT inhibition leads to mTORC1 inhibition. Thus, PI3K/AkT inhibition exhibits a broad spectrum of antiiflammatory events including NFkB inactivation, FOXO upregulation, HIF repression, etc.
(5) mTORC1 inhibition. Curcumin, resveratrol, EGCG, genistein, and caffeine inhibit both mTORC1 and mTORC2 [149], which is a consequence of AMPK activation and/or PI3K/AkT inhibition. mTORC1 inhibition leads to HIF repression, NF B inactivation, and autophagy upregulation.
There is also evidence for direct inhibition on mTORC1 by phenolic phytochemcals. Resveratrol significantly increases the association between mTOR and its negative regulator (DEPTOR), thereby downregulating mTOR activity [150]. Curcumin decreases total expression of mTOR, Raptor, and Rictor protein and mRNA levels [151]. Without affecting upstream kinase activities and TSC1/2 interaction, curcumin is also able to dissociate raptor from mTORC1 [152].
(6) FOXO activation. FOXO upregulation by phytochemcials largely results from PI3K/AkT inhibition and AMPK/SirT1 activation, which mediates antiinflammation (NFkB inactivation, HIFa repression, and Treg differentiation) and antioxidant enzyme expression. For cardioprotection per se, FOXO upregulation also shows anti-hypertrophic action by atrophic gene expression [10,11] as well as decreased ET-1 production.
(7) ERK inhibition. ERK inhibition is coherent with mTORC1 inhibition, presenting anti-inflammatory in addition to antiproliferative activities. ERK is at the upstream of mTORC1; ERK phosphorylates and inactivates TSC1 that negatively regulates mTORC1. Curcumin inhibits MAP3K/MAP2K and in turn MAPK (JNK, p38MAPK, and ERK) activation, thereby downregualting AP-1-mediated transcription activity for TNFa, iNOS, and COX-2 expression [145-148].
(8) JAK/STAT inhibition. Curcumin could inhibit JAK that otherwise phosphorylate STAT3, thereby blocking STAT3 dimerization and nuclear translocation. Such JAK/STAT3 attenuation exhibits anti-inflammatory (e.g., repressed proinflammatory genes) and anti-cancer (e.g., induced apoptotic proteins, suppressed apoptotic inhibitors, HIF1, VEGF, and c-Myc oncogene, etc.) activities [145- 148]. Similarly, resveratrol prevents JAK phosphorylation, thereby inhibiting STAT1 phosphorylation and transcriptional activity for IFN-related gene expression [153,154]. In a recent study, Noh et al. [155] have revealed that such JAK/STAT1 inhibition by resveratrol extends its benefit to indoleamine 2, 3-dioxygenase suppression for cancer immunoprotection.
(9) IKK/JNK inhibition. EGCG, for example, inhibits inflammatory serine/threonine kinase (e.g., JNK or IKK) directly or as a result of AkT inhibition to ensure antiinflammation [156]. In addition, such JNK/IKK inhibition attenuates insulin resistance; otherwise, JNK/IKK compete tyrosine phosphorylation on insulin receptor substrate (IRS).
(10) eNOS activation. Polyphenol-mediated AMPK activation phosphorylates at Ser1774 and activates eNOS [157]. As a consequence of antioxidation of upregulating SOD isoforms (SOD1- SOD3), glutathione peroxidase 1 (GPx1), and catalase, resveratrol downregulates NOX2 and NOX4, ensuring tetrahydrobiopterin availability and thus stabilizing eNOS activity. In addition resveratrol promotes eNOS mRNA expression. NO maintains EC integrity and VSMC relaxation, which is also considered anti-inflammatory. In an EC-dependent vasodilation, resveratrol activates eNOS activity, NO production, sGC induction, and subsequent cGMP production, completementing its ability to function as a non-selective PDE inhibitor (see the next section on PDE inhibition) for antihypertension.( 11) PDE inhibition. In fact, PDE inhibition by phytochemicals (e.g., resveratrol) precedes and contributes to AMPK activation [158]. As a non-selective PDE competitive inhibitor, resveratrol could exert diverse physiological effects in context of cyclic nucleotide involvements in biological events. It readily leads to the accumulation of cAMP and cGMP, both of which trigger ECindependent vascular dilation. Essentially, (a) cGMP induces Ca+2 effluxes, thereby leading to VSMC relaxation for antihypertension. (b) cAMP relaxation of rat aortic smooth muscle is mediated by (i) decreases in depletion of intracellular Ca2+ stores thereby reducing Ca2+ release and (ii) inhibition of capacitative calcium influx through store-operated calcium entry [158-161]. (c) PDE inhibition similar to PGI2 antiinflammatory mechanism favors cAMP accumulation and PKA activation, also exhibiting its anti-inflammatory potential.
(12) ß-catenin inactivation. By inhibiting PI3K/AkT, curcumin blocks axin/APC/GSK3ß /ß-catenin complex disassembly to cause ß-catenin degradation. Moreover, curcumin directly inhibits GSK3 for quenching -catenin release and nuclear translocation [162-164]. As a result of - catenin inactivation, transcription factors: PPAR and C/EBP are also downregulated.
(13) downregulation on TLR expression. To arrest inflammatory initiation and propagation, polyphenols such as curcumin, kaempferol-3-O-sophoroside, and EGCG inhibit TLR2/4 expression [165], which could reduce a wide-range of inflammatory responses such as LPS, IL-1 a/ß, and HMGB1 signaling.
(14) ACE inhibition. ACE inhibition by polyphenols presents several-fold significance in cardioprotection. As a consequrnce of suppressed AT-II formation, AEC inhibition essentially attenuates AT-II-mediated NOX activation, sGC inhibition, ET-1 elevation, thereby presenting not only anti-hypertension but also improved EC function as well as anti-inflammation. Most flavonoids are reported to be competitive inhibitors of ACE [166]. Anthocyanins, flavonols (e.g., quercetin, kaempferol, and myricetin), and flavanol (e.g., catechins, epicatechins, and their polymers) are effective ACE inhibitors. Flavonoid precursor molecules chalcones (butein) and their pyrazole derivatives also dose-dependently inhibit ACE in vitro. Methylated epigallocatechin-3-O-(3-O-methyl) gallate has shown more effective inhibition than its parent molecule epigallocatechin- 3-O-gallate. ACE inhibitory properties of flavones remain unclear; however, isoflavones (e.g., genistein, daidzein, and glycetin) decrease ACE gene expression and enzyme activity.
(15) attenuated ET-1 production and signaling. Nuclear exclusion of phosphorylated FOXO1 by EGCG results in downregulation of ET-1 promoter, thereby suppressing ET-1 expression and its activity [167]. Similarly, hydroxysafflor yellow A (HSYA), resveratrol, and quercetin reduce ET-1 production. In addition, green tea or EGCG downregulates ETA receptor, blocking ET-1 signaling for anti- hypetension and hypertrophy.
(16) adiponectin elevation and signaling. Increased adiponectin is reported in response to resveratrol that also upregulates the expression of adiponectin receptor-1. Resveratrol promotes the posttranslational multimerization and stability of adiponectin by DsbA-L that is induced upon AMPK activation and AkT-mediated FOXO1 activation [168]. In a clinical trial [169], grape resveratrol increases serum adiponectin and downregulates inflammatory genes (PAI-1, IL-6, AP-1, JUN, CREBP, etc.). In an animal model [170], 7-O-galloyl-D-sedoheptulose increases adiponectin level while downregulating leptin, insulin, C-peptide, resistin, TNFa, and IL-6 in serum and proinflammatory NFkB p65, COX-2, iNOS, JNK, phospho-JNK, AP-1, TGFß1, and fibronectin.
(17) K+ channels activation. In an EC-independent fashion for vasodilation, resveratrol directly opens K+ channels including K+ ATP and BK+ Ca expressed on VSMC where extracellular Ca+2 influx and intracellular Ca+2 release are also suppressed [160].
(18) autophagy upregulation. AMPK activating signals are able to upregulate autophagy, which is mediated by mTORC1 inhibition. MTORC1 negatively regulates a complex consisting of essential autophagic proteins (e.g., ULK1, ATG13, ATG101, and FIP200). Alternatively independent of mTORC1 inhibition, direct PI3K/ AkT inhibition could block phosphorylation of a crucial autophagic element: Beclin 1 that otherwise dimerizes and recruits 14-3-3 further being sequestered to cytoskeletal actin vimentin and intermediate filament complex. As a result of such blockage mediated by PI3K/AkT inhibition, autophagosome assembly is able to initiate autophagy.
Specifically, the proposed biochemical mechanisms involve (a) DAPK phosphorylates Thr119 in the BH3 domain of Beclin 1, preventing Beclin 1 interaction with Bcl-2/Bcl-x, or (b) ULK1 or JNK phosphorylates Beclin 1 inhibitor Ambra1 or Bim, respectively, thereby preventing their interactions with Beclin 1 and its consequent sequestration to microtubule Dynein. The resulting released and inactive free Beclin 1 is available for the assembly of autophagosome. For instance, EGCG increases formation of LC3- II and autophagosomes, downregulating ectopic lipid accumulation through such increased autophagic flux.
Essentially, autophagy, an intracellular cleaning system including aggrephagy, xenophagy, mitophagy, and lipophagy, contributes to regenerating metabolic precursors, and cellular and tissue homeostasis by degrading long-lived proteins, protein aggregates, and defective organelles (e.g., mitochondria, ER, or peroxisomes), and cleaning subcellular debris.
Autophagy prevents inflammation (e.g., NLRP3 inflammasome activation). (1) Mitophagy limits NLRP3 activation by removing damaged mitochondria. (2) Autophagosomal Atg16L1 readily inhibits ROS production; ROS is essential for NLRP3 activation. (3) Autophagy per se promotes lysosome (NLRP3 inflammasome) degradation through ubiquitination involving autophagosomal components p62 and LC3. (4) Furthermore, removal of pro-IL1ß /18 by autophagy for NLRP3-mediated caspase 1 cleavage ensures antiiflammation. Thus, autophagy protects from inflammasome (NLRP3) activation that is essential for IL-1ß /18 maturation and secretion. Consistently, defective autophagy is widely associated with inflammation, neurodegenerative diseases (e.g., Alzheimer’s disease), autoimmunity (e.g., Crohn’s disease, systemic lupus erythematosus), cancers, infection (e.g., TB), aging, etc.
Limited information is available directly regarding autophagy upregulation in relation to cardioprotection. Apparently, resveratrol induces autophagy and thus possibly protects from MI.
Other biological actions
(1) Inhibition on a-glucosidase. Most phytochemicals flavonoids, curcumin, alkaloids, anthocyanins, terpinoids, and others are -glucosidase inhibitors [171]. a-Glucosidase activity in GI brush border of the small intestines is responsible for glucose generation from non-fiber oligo-or poly- saccharides. Such inhibition limits glucose inputs from dietary carbohydrate digestion and absorption, certainly contributing to caloric restriction, reduction of hyperglycemia, and lowered diabetes risk.
(2) Upregulation on paraoxonase 1. Quercetin increases paraoxonase 1 mRNA and protein expression, upregulating paraoxonase 1 activity. Paraoxonase 1 is a HDL-associated enzyme displaying esterase and lactonase activity; paraoxonase 1 metabolizes toxic OxLDL or OxHDL, thus protecting LDL and HDL from oxidation [172].
(3) Anticoagulation. Significantly prolonged TT, aPTT, and PT have been reported in vitro, in vivo, or ex vivo in response to polyphenols. (a) Thrombin inhibition [173,174]. Curcumin and its derivative bisdemethoxycurcumin, cyanidin, quercetin, silybin, cyanin, (+)-catechin and (-)-epicatechin inhibit thrombin amidolytic activity; in addition, cyanidin, quercetin, and silybin suppress thrombin proteolytic activity. Aglycones act as competitive thrombin inhibitors, while chokeberry extract significantly inhibits thrombin amidolytic activity. (b) FXa inhibition [174-176]. Flavonoids: procyanidin B2, cyanidin, quercetin, and silybin bind S1-S4 pockets in vicinity of the FXa active site and block access of substrates to Ser195, thereby directly inhibiting FXa amidolytic activity. Curcumin and its derivative bisdemethoxycurcumin also inhibit FXa activity. (c) Protection from FVII activation [177]. Tannic acid, delphinidin, hamamelitannin, (-)-epicatechin gallate, and 3,5-di-O-caffeoylquinic acid bind plasma hyaluronan-binding protein and inhibit FVII autoactivation (autoproteolysis). (d) TF suppression/encryption [178]. Grape and its products with high content of polyphenols exert anticoagulation by suppression of TF synthesis in blood mononuclear cells and VECs. In a recent personal communication, antiinflammatory HSYA (a phenolic related flavonoid component from Carthamus tinctorius L.: 3,5,6-trihydroxy-2(E)-[1-oxo-3- (4-hydroxyphenyl)-2-propenyl]-4,6-bis[(2S,3R,4R,5S,6R)-3,4,5- trihydroxy-6-(hydroxyl-methyl) oxan-2-yl]-2,4 cyclo-exadien-1-one) suppresses OxLDL-induced TF expression in vitro/in vivo models, which is mediated by PPARγ upregulation and attenuated p38 MAPK phosphorylation/activation. Rutin (flavonoid) has long been recognized as an anticoagulant for possible prevention of heart attack and stroke, which is mediated by its inhibition on protein disulfide isomerase that de-encrypts TF for initiating the extrinsic coagulation pathway and robusting thrombin formation. (e) Other actions. Aronia melanocarpa or seeds of Vitis vinifera prolong clotting time and decrease the maximal velocity of fibrin polymerization and FXIIIa amidolytic activity in human plasma [179]. However, there is no evidence thus far whether polyphenols have effects on natural anticoagulants (e.g., TFPI, APC, or AT III).
(4) PAI downregulation. By stimulating binding of upstream stimulatory factor-2 to two distinct E-box sequences, quercetin downregulates PAI-1 promoter, thus resulting in suppressed PAI- 1 expression in human coronary artery ECs [180]. Polypenols including curcumin, quercetin, resveratrol, and EGCG and its derivatives (octaacetate and theaflavin digallate) act as potential PAI- 1 inhibitors to reduce PAI-1 production [181], favoring fibrinolysis and resolution of blood clots. Similarly, grape ingradients suppress PAI-1 levels [182,183].
(5) tPA upregulation. Quercetin induces tPA expression, which is mediated by functional Sp1-binding element in tPA promoter and p38 MAPK pathway [184]. Catechin, epicatechin, quercetin, and resveratrol in red wine induce TPA and u-PA in vitro [185].
(6) Altered gut microbiota. As a prebiotics, pholyphenols could alter the landscape of gut microbiota [186-188]. For instance, resveratrol supplementation suppresses Parabacteroides johnsonii, Alistipes putredinis, and B. vulgatus induced by high-fat, which is proposed to enhance GLP-1 secretion [189]. Pomegranate extract rich in Gallic and ellagic acid enhances the total growth of Bifidobacterium spp. and Lactobacillus spp. without affecting C. coccoides-E. Rectale and the Clostridium histolyticum groups [190]. Green tea [191] increases the survival of Bifidobacteria, known probiotics.
(7) Epigenetic modulations. EGCG inhibits DNA methyltransferase activity in various experimental and clinical studies [192,193]; epigenetic modulations could offer cardioprotection.
Cardioprotection by Polyphenols
Figure 1 depicts antagonisms by polyphenols against CVD risks. The “spider web-like” multiple effects offer cardioprotection by antioxidation, AMPK activation, eNOS activation, SirT1 activation, ACE inhibition, PDE inhibition, hypolipidemic effects, anti-coagulation, anti-platelet aggregation, fibrinolysis, improved EC function, reduced food intake/weight gain, and suppressed lipogenesis from in vitro, animal models, and clinical trials.
There are significant cross-talks and enhancement among polyphenolic biological actions. For instance, polyphenol-induced GLP-1 secretion (not shown) largely resulting from gut microbiota modification forms a positive feed-forward loop for ensuring cardioproteiction (e.g., atherosclerosis, hypertension, fibrillation, arrthymia, HF, etc.) [194], which essentially reinforce AMPK activation, NO bioavailability, natriuretic peptide secretion, cAMP/ cGMP upregulation, improved EC function, vasodilation, antiinflammation, etc. In addition to its anti-hypertension including EC- dependent and independent vasorelaxation and decreased kidney sodium reabsorption, GLP-1 provides anti-diabetic actions of antagonisms against insulin resistance and glucose intolerance.
Antagonisms against CVD risks
(1) Anti-oxidative stress. (a) The classical antioxidation of polyphenol presents anti-oxidative stress. Curcumin [145-147] readily scavenges free radicals with upregulated antioxidant enzymes (catalase and MnSOD [148]) and reduced OxLDL. Flavanol reduces copper ion-mediated LDL oxidation [195]. Quercetin inhibits LDL oxidation with elevated paraoxonase 1 [172] while removing atherogenic lesions and lipoprotein oxidized lipids [196]. Similarly, resveratrol suppresses LDL oxidation. (b) ACE inhibition by polyphenols is also in line with such antioxidative effects in view of AT II formation in contribution to ROS production and inflammation [29]. (c) The anti-inflammatory actions of polyphenols include NFkB/AP-1 inactivation and HIF repression, which in turn blocks ROS generation. Such anti-oxidative stress also readily contributes to anti-inflammation.
(2) Hypolipidemic effects. The ability of polyphenols to activate AMPK could largely account for hypolipiemic effects; AMPK, a classical energy sensor, favors catabolism for energy production. AMPK historically recognized to phosphorylate HMG-CoA reductase and ACC inhibits de novo biosyntheses of cholesterol and fatty acids and TG formation. Other consequent downregulations on lipogenic gene and enzyme expression further reinforce AMPK hypolipidemic actions; polyphenol-induced AMPK-mediated mTORC1 inhibition readily offsets mTORC1-activated lipogenic transcription factor: SREBPs. Curcumin’s hypolipidemic effects include suppressed SREBP1/2, CREB, CREBH, PPARγ, and LXRa, while lipid homeostasis results from dowregulated aP2/FABP4, CD36, HMG-CoA reductase, and carnitine palmitoyltransferase-I [145-147]. Flavanone derivatives (e.g., hesperetin) in orange juice appreciably lower LDL/HDL ratio. Coffee consumption increases serum adiponectin and decreases total serum cholesterol, HDL-C, and ApoA-I with decreased ratios of LDL-C/HDL-C and ApoB/ ApoA-I. Olive oil polyphenols decrease LDL concentrations and LDL atherogenicity [197].
As a consequence of AMPK activation and phosphorylation, polyphenols could exhibit hypotrigleridemic action by increasing LPL activity and decreasing ANGPTL4 mRNA expression [198]. Similarly, apple polyphenols upregulate LPL activity in an animal model [199]. (-)-Epicatechin also significantly decreases hyperTG [200].
(3) Improved EC functions. (a) Polyphenols are capable of suppressing ACE and PDE activities. (i) ACE inhibition becomes a focal event in improving EC function. By blocking AT-II generation and AT-II-mediated endothelial dysfunction including oxidative stress (NOX activation), ET-1 elevation, and sGC inhibition, ACE inhibition readily respectively promotes NO bioavailability, lowers Ca+2 influx/[Ca+2]i, and cGMP generation, thus in turn leading to vasodialation. (ii) PDE inhibition leads to improved EC function in addition to anti-inflammation in concert with NO-independent cAMP/cGMP accumulation as well as EC-independent VSMC relaxation, which complements ACE inhibition for anti-hypertension. (b) As a consequence of AMPK activation and FOXO1 upregulation, ET-1 suppression by polyphenols also improves EC function [167]. For instance, HSYA reduces ET-1 production, NOS expression, and oxidative stress, protecting rats from acute and chronic congestive cardiac failure.
(4) Anti-thrombosis. Polyphenols’ anticoagulation, fibrinolytic, and anti-platelet functions readily ensure anti-thrombotic activities. (a) Polyphenols generally activate AMPK and eNOS, in turn enhancing NO bioavailability; NO protects platelets from activation and aggregation [43]. Such increased NO bioavailability with S-nitrosylated activated tPA could possibly provide antithrombosis. (b) The classical antioxidative potentials to suppress ROS production improve EC function and NO bioavailability. (c) The direct anticoagulation (thrombin inhibition [173, 174], FXa inhibition [174-176], protection from FVII activation [177], TF suppression/ encryption [178], downregulated fibrin polymerization and FXIIIa [179]) complemented by clot resolution (e.g., PAI-1 downregulation, tPA upregulation) confers antithrombosis. (d) The anti-inflammatory potentials to interrupt the coagulation-thrombosis-inflammation circuit [48,49] could lead to anti-thrombosis. (e) Among antiinflammatory actions, NF B inactivation and consequent COX inhibiton resulting in TxA2 suppression are in line with anti-platelet.
Resveratrol reduces platelet aggregation. Aronia melanocarpa with an extremely high content of procyanidins and anthocyanins reduces platelet adehesion and aggregation [201], while flavanol and procyanidin dimers attenuate platelet reactivity [195]. Similarly, quercetin and apigenin inhibit platelet secretion and aggregation. Ellagitannins are of vascular health including anti-atherogenic, anti-thrombotic, anti-inflammatory and anti-angiogenic effects while reducing copper ion-mediated LDL oxidation [196]. EGCG inhibits COX-1, TxA2 production, and TxA2 signaling, which confers antagonisms against thrombotic disease-associated platelet aggregation. In view of platelet activation/aggregation being proinflammatory, such phenolic actions are also anti-inflammatory. HSYA could significantly reduce platelet aggregation and protect VECs.
(5) Anti-hypertension. Both AMPK- dependent and independent polyphenolic actions confer anti-hypertension that is either EC- dependent or independent. (a) ACE and PDE inhibition mainly contribute to anti-hypertensive actions independent of AMPK activation. (i) By targeting RAAS, polyphenol-induced ACE inhibition readily reduces AT-II production and AT-II-mediated consequences in oxidative stress, sGC inhibition, and endothelial dysfunction. (ii) The suppressed AT-II-mediated sGC inhibition synergistic with PDE inhibition is in favor of cGMP bioavailability for vasodilation. (iii) The improved EC function including reduced ET-1 production readily exhibits EC-dependent relaxation. (b) Specifically, polyphenol-induced AMPK activation strengthens cardioprotection and maintenance, which is mediated by phsophorylating/ upregulating eNOS and its consequence of cGMP/PKG upregulation. (c) Furthermore, the classical antioxidative potentials of polyphenols suppress, for instance, O2•- formation and in turn increase NO bioavailability. Enhanced NO bioavailability and sGC activation are responsible for cGMP enrichment, Ca+2 efflux, and VSMC relaxation. Alternatively, PKG phosphorylates myosin phosphatase to actually dephosphorylate myosin, promoting contractile. (d) In EC-independent fashions, the direct activation on K+ channels blocks [Ca+2]i increases, causing VSMC relaxation [160]. Direct PDE inhibition by polyphenols favors cGMP and cAMP accumulation for vasodilation.
Anti-hypertensive potentials of resveratrol are attributed to ACE and PDE inhibitions [141,142] and eNOS upregulation [141-143,202- 204]; thus, cGMP accumulation and Ca+2 effluxes ensure vascular dilation [160,201]. Resveratrol [142] also inhibits cAMP-degrading PDEs (e.g., PDE4), resulting in accumulated cAMP, a vasodilator. Similarly, anthocyans exert anti-hypertension by eNOS activation and ACE inhibition [205,206]. Flavanol increases NO bioavailability for EC-dependent vasodilation [195]. HSYA involving in activation of BK(Ca) and K(ATP) channels could significantly reduce blood pressure and heart rate.
(6) Anti-inflammation. (a) Polyphenols target multiple inflammatory components by antioxidant potentials, AMPK activation, PI3K/AkT inhibition, IKK/JNK inhibition, mTORC1 inhibition, JAK/STAT inhibition, suppressed HMGB1 and granzyme B release, and TLR suppression. As a result, polyphenols readily lead to NFkB, AP-1, HIF, and STAT1/3 inactivations with reduced proinflammatory mediators (e.g., PGE2, cytokines, adhesion molecules, growth factors, etc.). (b) Polyphenol-mediated ACE inhibition and its antagonisms against AT-II-mediated consequence in oxidative stress could contribute to anti-inflammation. (c) Polyphenol-induced anticoagulation [173-179] and anti-platelet aggregation [201] (e.g., COX inhibition; reduced TxA2) could arrest the coagulation-thrombosis-inflammation circuit [48,49], showing anti-inflammation. (d) Polyphenols also sustain resolution of inflammation (e.g., SirT1 activation, eNOS activation, FOXO upregulation, PDE inhibition, and adiponectin elevation), which reinforces anti-inflammatory actions for promoting, restoring, and maintaining tissue homeostasis [77]. (e) Autophagy upregulation by polyphenols prevents NLRP3 inflammasome activation, a characteristic cellular inflammation.
For instance, capsaicin represses expression of pro-inflammatory gene iNOS and COX2 activity and PGE2 production through NFκB inactivation by blocking IkBa degradation, reduced JNK activation, and inhibition on the production of cytokines (e.g., TNF-a, IL-6 and MCP-1) [207-209]. Capsaicin suppresses TNF release by blocking TNF mRNA transcription resulting from dampened NFκB binding to the DNA promoter (personal communication). Further, capsaicin inhibits Mφ activation, downregulates TLR-2/4 [165], transcription factors (e.g., NFκB, AP-1, STAT) and enzymes (e.g., COX-2, LOX, MMP9, MAPK, mTOR, AkT, IKK, c-jun/fos), reduces the expression of inflammatory mediators (e.g., PGE2, LTs, cytokines (e.g., TNF, IL-1), and adhesion molecules (e.g., ELAM-1, ICAM-1, VCAM-1)) [145-147,208], and upregulates PPAR [210]. The phenylethyl amide of caffeic acid decreases TNFa and inactivates NFκB, while caffeic acid phenethyl ester inhibits 5-LOX for LT biosynthesis, blocks IL-6-induced STAT3 activation, and suppresses TLR-4 activation and LPS-mediated NFκB and IRF3 activation in Mφs [211,212]. Gingerol attenuates COX-2 expression by blocking p38 MAPK, NFκB activation, and TNF-a expression. Procyanidins reduce the expression of IL-6 and MCP-1 while enhancing the production of the anti-inflammatory adiponectin [204,208]. Procyanidin B2 inhibits NLRP3 inflammasome activation in human vascular endothelial cells, which could offer vascular protection. EGCG blocks NFκB activation in human ECs and thereby inhibits MCP-1 expression. Interference with MAPK pathways and the down-modulation of iNOS transcription and NO production in MFs further ensure anti-inflammatory potentials of EGCG [207,213,214]. EGCG analog (piceatannol) inactivates NF B and inhibit JAK-1 for blocking COX-2 expression and action [207,215]. Quercetin decreases inflammation markers (IFN-γ, IL-1 a/β 1/4/6/8, and MCP-1 expression), inhibits JNK- and ERK - phosphorylation and activation, and prevents AP-1 and NFκB activation. Furthermore, quercetin, apigenin, and genistein interfer with thrombin intracellular signal through PAR4, which is proposed to be mediated by calcium mobilization. HSYA upregulates PPARγ, inactivates AP-1/NFκB, and suppresses MAPK activation/ phosphorylation for anti-inflammation.
(7) Anti-diabetes. (a) Polyphenols reinforce physiological insulin action with additional protection from insulin resistance [216-218]. (i) AMPK activation shows positive effects on eNOS that leads to increased glucose uptake/utilization, which mimics not only muscle contraction [219] but also the mechanism by which insulin works in vivo [211,212]. eNOS activation generally accounts for enhanced insulin action with increased glucose uptake by peripheral tissues via Glut-4 [143,203,204]. In addition, such eNOS activation generating NO mediates Glut4 translocation and increased glucose uptake/utilization by muscle cells [203,204], mimicking insulin action. (ii) AMPK-induced adiponectin elevation [168] exerts antiinflammation and insulin sensitivity. (b) The polyphenol-induced PI3K/AkT inhibition accompanying with JNK inhibition could lead to suppressed insulin resistance. (c) mTORC1 inhibition resulting from either AMPK activation or PI3K/AkT inhibition consistently promotes glycolysis (glucose utilization). (d) a-glucosidase inhibition by polyphenols [220] reduces glucose inputs from dietary carbohydrates, certainly lowering hyperglycemic risk. (e) Polyphenol (resveratrol) increases GLP-1 production [189] showing enhanced insulin and decreased glucagon secretion and conferring anti-diabetic action. Moreover, the ability to inhibit the activity and expression of DPP-4 could sustain GLP-1 bioavailability and benefit to anti-diabetes (personal communication) synergistically with upregulated GLP-1 receptor on cells. GLP-1 is known to mimic insulin action in addition to increasing insulin secretion, suppressing apoptosis, and enhancing differentiation. (f) Recent insights reveal that flavonoids are able to enhance insulin secretion by promoting proliferation while reducing apoptosis of pancreatic β-cells. (g) As prebiotics, polyphenols could alter gut microbiota which contribute to energy harvesting from diets, satiety, insulin sensitivity, etc. [80]. Resveratrol significantly improves insulin sensitivity and decreases insulin resistance. As a consequence of activation of the AMPK-SirT1-PGC1a axis [142,141], resveratrol readily reduces blood glucose, insulin levels, liver fat storage, and inflammation while improving muscle mitochondrial function. Gingerol inhibits JNK activation, thereby protecting insulin signaling from IRS serine/threonine phosphorylation (i. e., insulin resistance). Similarly, anthocyans reduce diabetes incidence through modulation of insulin sensitivity and glucose utilization. (-)-Epicatechin significantly decreases glycemia [221]. HSYA in vitro shows antiglycation, attenuating AGE formation.
(8) Anti-obesity. Polyphenols show the abilities to suppress lipogenesis and adipogenesis, which generally shares with antiobesity actions [222]. (a) AMPK activation inhibits HMG-CoA reductase (cholesterol synthesis), ACC (fatty acid synthesis), and TG formation. For instance, resveratrol and quercetin combination [223] clinically shows ACC inhibition and adipose triglyceride lipase upregulation without any effect on HSL. (b) Its consequent SirT1 activation ensures the inhibitions on the genes involved in adipocyte differentiation and TG accumulation. As a consequence of mTORC1 inhibition, suppressed ADD1/SREBP-1c signals are associated with decreased levels of PPARγ as well as C/EBP a/d mRNA levels during adipogenesis [209,222], thus offering anti-obesity. (c) AMPK activation also mediates adiponection expression that leads to foodintake suppression and weight loss in addition to anti-inflammation. (d) As prebiotics, polyphenols could alter gut microbiota with suppressed “obese microbiota” [80].
Capsaicin [208,224] inhibits adipogenesis/adipocyte differentiation, lowers intracellular TG, and enhances BAT thermogenesis. By stimulating lipolysis and thermogenesis, capsaicin thus increases the energy expenditure in adipose tissue. For activating brown adipose tissue, upregulation of mitochondrial UCP-2 prevents adipogenesis and obesity. Its stimulation on lipid mobilization from adipose tissue, increased fat oxidation, and lowering the perirenal adipose tissue weight and serum TG readily present anti-obesity. Clinically, capsaicin increases satiety and reduces energy intake and hunger [208,209,224]. By inhibiting JNK phosphorylation, gingerol similarly upregulates adiponectin expression, thereby accounting for satiety and weight loss.
EGCG’s anti-obesity effects include downregulation of the expression of major adipogenic genes (e.g., PPARγ, C/EBPa, FABP4, and FAS) involved in adipogenesis [207,210,213]. In addition to inhibiting PI3K/AkT pathway, EGCG inactivates MEK/ERK pathway, resulting in activation of FOXO1 transcription activity and inhibiting preadipocyte differentiation to mature adipocyte [222,225]. Phenolic honokiol from mango increases plasma adiponectin level. (-)-Epicatechin [200] significantly decreases hypertriglyceridemia and the rate of weight gain while restoring SirT, PGC-1a, mitofilin, TFAM, and UCP1 in animal models.
(9) Modulation on hyperhomocysteinemia. Limited confirmatory information is known about the direct effects of polyphenols on homocysteine level. However, diverse polyphenolic actions on antioxidation, AMPK activation, eNOS phosphorylation/ activation, inactivation of PI3K/AkT/mTORC1 and MAPK/ERK signaling, anticoagulation, PAI-1 reduction, ET-1 attenuation, anti-platelets, etc. could be expected to significantly counteract hyperhomocysteinemia consequence in CVD (see the section of Hyperhomocysteienemia under Major CVD risks). For instance, chokeberry, resveratrol, and grape seed extract reverse homocysteineinduced platelet adhesion and aggregation [179], while aronia melanocarpa fruits (e.g., rosaceae) suppress homocyteine-induced hemostatic effects [226].
Benefits to CVD: In summary, polyphenols could offer a broad spectrum of benefits to CVD (Figure 2). Among many different CVD forms, they often result from commonly shared risks including oxidative stress, endothelial dysfunction, inflammation, and hypertensive and thrombotic features. In CVD vicious network, the disease types often manually enhance and refuel one another. In animal models and clinical trials, the abilities to activate AMPK, interrupt intracellular signaling of CVD development, and inhibit ACE and PDE could readily make polyphenols versatile in cardioprotection in addition to their antioxidative properties.

Figure 2: Multiplebenefits of polyphenols protect against CVD. The left panel depicts major polyphenolic actions that could multiply ease CVD. The right panel
show major forms of CVD with key contributing characterisitics highlighted in parentheses. Please refer to the text for details. ↑ denotes upregulation, activation, or
increased production, while ↓ denotes downregulation, inhibition, inactivation, or suppressed expression/production.
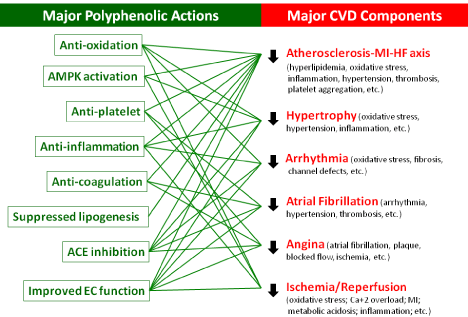
Graphical Abstract:
(1) Protection from atherosclerosis, MI, and consequences. Polyphenols target several key atherogenic components including endothelial dysfunction, hyperlipidemia, oxidative stress, inflammation, and hypertensive and thrombotic events. For instance, coca products suppress LDL oxidation in hypercholesterolemia rabbits, while the antiatherogenic activity of anthocyans is mediated by inhibition of NFκB-mediated VCAM/ICAM expression and upregulation on antioxidative enzymes (e.g., Nrf2-ARE activation) [227,228]. EGCG activates autophagic system/flux, thereby showing downregulation on lipid accumulation.
NOX2/4 upregulation plays a pathogenic role in cardiac ischemic as well as reperfusion phases in I/R injury [229]. Accordingly, the ability to inhibit NOX by polyphenols could be of protection; resveratrol readily prevents the heart from I/R injury [230]. In conjunction, there is reduced NO bioavailability due to ROS (e.g., O2•-) overproduction; reduced NO presents endothelial dysfunction and vasoconstrinction, in turn manifesting as CVD risks. AMPKmediated eNOS activation by polyphenols complements such deficit and provides protection from I/R injury and its progression to MI and HF. Similarly, S-nitrosylation of L-type Ca2+ channels by eNOSderived NO protects from I/R injury. In addition, resveratrol-induced autophagy by either AMPK-mediated mTORC1 inhibition or PI3K/ AkT inhibition offers cardioprotection from myocardial ischemia. In addition to anti-oxidant effects, HSYA suppresses TLR4 signaling and AkT/hexokinase II independent of ERK/GSK-3β pathway for protecting I/R injury in experimental models.
(2) Protection from hypertrophy. Polyphenols readily provide antagonisms against hypertrophy. (a) Mechanistically, polyphenols could attenuate hypertrophic pathogenesis by FOXO upregulation (atrophic gene expression), PI3K/Akt/mTORC1 inhibition, -catenin inactivation, and downregulating [Ca+2]i and its consequent calcineurin-dependent NFAT activation. In addition, polyphenol’s anti-inflammatory potential with NFkB inactivation attenuates hypertrophic gene expression. (b) Anti-hypertrophic actions could be achieved by EC-dependent or independent VSMC relaxation (see the above section on Antagonism against CVD risks; (5) antihypertension by polyphenols). (c) Anti-thrombosis including direct anti-coagulation could complement anti-hypertrophic actions. (d) The ACE inhibition and its consequences (e.g., anti-hypertension, reduced PAI-1/2, ET-1 attenuation, anti-oxidation, and anti-platelet adhesion/aggregation) could show anti-hypertrophy. (e) The direct downregulated expression of ET-1 and ETA receptor by green tea or EGCG [231] is responsible for improved EC function and antihypertrophic antihypertrophic benefits. (f) The classical antioxidant potentials of polyphenols could readily offset a key role of ROS in hypertrophy [13,14].
EGCG inhibits hypertrophy by antioxidation and NFkB/AP-1 inactivation [232]. Resveratrol activates AMPK and eNOS [233,234] and suppresses ET-1 expression [235], achieving anti-hypertrophy. Resveratrol reduces hypertrophic growth of the myocardium, which could be mediated by reduced hemodynamic load and inhibition of the p70 S6 kinase pro-hypertrophic signaling cascade [236] in addition to its antioxidative, antihypertensive, and improved vascular functions. Curcumin inhibits nuclear localization and DNA binding activity of GATA-4 to show anti-hypertrophic potentials [237]. The p300-HAT inhibitory effects of curcumin reduce the development of cardiac hypertrophy and HF in animal models [238]. By preventing telomere shortening accompanied by the loss of telomere repeatbinding factor, EGCG, quercetin, and carvedilol are reported to inhibit cardiac myocyte apoptosis [239], a component in hypertrophy. In animal models, resveratrol arrests and regresses the development of pressure overload-induced cardiac hypertrophy. HSYA attenuates LV remodeling after pressure overload-induced cardiac hypertrophy.
(3) Protection from arrhythmia and consequence. Little is known about direct protection of arrhythmia by polyphenols. The abilities of polyphenols to inhibit ACE, PDE, blood coagulation, PAI-1 production, and platelet aggregation could contribute to anti-arrhythmia. The ACE inhibition reduces fibrosis, while the anticoagulation shows reduced risk. K+ channel activation and downregulation of [Ca+2]i by polyphenols [160] could also be expected to exhibit anti-arrhythmia. In view of oxidative stress in arrhythmia [15-17], the classical anti-oxidative potentials of polyphenols could certainly provide antagonisms against arrhythmia and AF, while the polyphenolic anti-thrombotic effects could also offer such benefits to AF. Angina, a type of chest pain caused by reduced blood flow to the heart muscle, is recognized as a major symptom of AF; accordingly, polyphenols could ease angina.
ECG, as a novel sodium channel agonist, could have clinical applications on cardiac arrhythmias [240]. Curcumin modulating [Ca+2]i homeostasis may play a role in the prevention of ventricular arrhythmias [238]. Coffee could reduce risks for both atrial and ventricular arrhythmias.
(4) Other CVD benefits. Flavanol-rich chocolate improves vascular and platelet functions by increasing NO bioavailability and decreasing oxidative stress, which eases congestive HF. By improving survival, EC-dependent smooth muscle relaxation, cardiac contractile, and mitochondrial function, resveratrol protects hypertension-induced HF in animals. Polyphenols in blueberries prevent cardiomyocyte death by preventing calpain activation and oxidative stress, protecting against hypertrophy and HF. Polyphenolinduced eNOS-driven S-nitrosylation of GRK2 inactivates GRK2 (a kinase responsible for β-adrengenic receptor activation and cell death), thus conferring protection from cardiomyocyte death and HF. HSYA reduces ET-1 production, NOS expression, and oxidative stress, protecting rats from acute and chronic congestive cardiac failure.
In an immunological view of cardioprotection from atherosclerosis, hypertension, MI, HF, etc. [241], the ability of polyphenols to induce immunosuppressive Tregs for anticancer activity [242] could similarly facilitate their antagonism against CVD.
Perspectives
A book entitled “You Are What You Eat: how to win and keep health with foods” published in 1942 addresses the link between a healthy diet and better health (recently refer to as naturopathic medicine; nutraceutics), which has been originally written by a nutritionist Henry Lindlahr (1862-1924). In a broader view, food could function as hormones; nutrients deriving from food can modulate intracellular events to regulate metabolic health by directly and indirectly activating receptors and signaling pathways. Nutrition in critical illness is also widely recognized [243], for instance, boosting immunity, fighting severe skeletal-muscle wasting and weakness, and supporting wound healing and recovery.
Cardioprotection by natural components
Figure 1 (right panel) demonstrates major polyphenolic actions in multi-targeting cell signaling and functions, which prevents CVD risks at many dimensions by antagonizing against oxidation, thrombosis, hypertension, obesity, diabetes, inflammation, endothelial dysfunction, etc. (Figure 1; left panel). Its anti-oxidation, AMPK activation, and diverse effects on downstream cell signaling pathways certainly extend health benefits beyond cardioprotection. Daily consumption with polyphenol-rich fruits and vegetables could be expected to promote human health.
Dietary ingredients such as n-3 PUFAs and fibers as well as many others have also been recommended for cardioprotection by lowering lipidemia and improving EC functions. For instance, phytosterols suppress dietary zoosterol absorption and lower serum cholesterol level. n-3 PUFAs (e.g., EPA and DHA) reduce CVD risks and ease myocardial damages, ventricular arrhythmias, atherosclerosis, thrombosis, ischemic heart disease, MI, and HF processes [244-246]. n-3 PUFA effects on EC function could be a major mode of action. n-3 PUFA is able to inhibit the expression of adhesion molecules (e.g., ICAM, VCAM and E-selectin), proinflammatory cytokines (e.g., TNF and IL-1/2), thrombomodulin, vWF, and PAI-1. As a result of promoting NO production, n-3 PUFA improves vasodilation. In addition, n-3 PUFAs exhibit potent hypotriglyceridemic action [244], raising HDL-C, small reduction in blood pressure, depressed platelet aggregration, antiinflammation, competitive inhibitors for AA metabolism, and reduced susceptibility of LDL to oxidation [245,246]. In recent developments, n-3 PUFAs lower circulating PCSK9 level as well as Idol expression accompanied with LDLR abundance in animal models and in vitro. There is a perception that dietary fibers generally delay and lower fat absorption. In addition, SCFAs (e.g., butyric acid) derived from colonic microbiota (e.g., Lactobacterium) fermentation of dietary fibers are anti-inflammatory [247] evidenced by Treg expansion and HDAC inhibition. SCFAs could also trigger anorectic incretin hormone (leptin, PYY, and GLP-1) secretion, which could account for the roles of gut microbiota and fibers in satiety, antidiabetes, anti-obesity, and cardioprotection.
Nuts are rich in fibers, high quality proteins, and minerals (e.g., Mg+2), which is shown to reduce cardiovascular mortality [248]. For instance, MgCl2 is proposed to be an inhibitor of HMGCoA reductase, which could present an effect similar to statin on cholesterol lowering for caridoprotection.
Moving forward to nutraceutics or therapeutics
Natural ingredient/product readily serves as a platform for drug developments. Good examples include compactin or mevastatin derived from fungi extracted by Endo group in 1976 [249]. Till now, phytochemical aspirin [250] remains mostly recommended for cardioprotection; the American Heart Association strongly advocates low dose aspirin (e.g., 80 mg) daily for healthy hearts. Its classical COX1 inhibition arrests PGE2 and TxA2 production; PGE2 is proinflammatory, while TxA2 is responsible for vasoconstriction and platelet aggregation. Such common blood-thinner aspirin essentially activates AMPK and promotes resolvin (Rv) formation from n-3 PUFAs [77,124], which presents a wide range of antiinflammation. In transcellular metabolisms, COX-2 acetylation by aspirin undergoing conformational changes leads to 18R-HpEPA or 17R-HpDHA formation. The R form derivatives are continually metabolized by 5-LOX and epoxide hydrolase in neutrophils; thus, RvE1 and at-RvD1 are formed from EPA and DHA, respectively. In a close relation to cardioprotection, RvE1 reduces ADP-stimulated platelet aggregation, TxA2 generation, P-selectin mobilization, and actin polymerization in a calcium-independent manner; RvE1 counter-regulation of ADP activation is ChemR23-dependent [251]. Furthermore, acetylated COX-2 catalyzes AA conversion to 15(R)- HETE that then through 5-LOX reaction followed by hydrolysis results in epi-LXA4 and epi-LXB4 derivatives, two anti-inflammatory mediators. Beyond cardioprotection, aspirin has also been used for diabetes, colorectal cancer, inflammation, Alzheimer’s, and many other medical applications.
Concerning biochemical actions, polyphenol-induced AMPK activation is analogous to anti-inflammatory (e.g., aspirin, methotrexate, pemetrexed, acadesine, and salicylate) [252] and antidiabetes (e.g., metformin, phenformin, and A769662) agents [253]. Direct and indirect mTORC1 inhibition could make polyphenols comparable to rapamycin benefits in inflammatory and aging related pathological conditions such as cognition decline, Alzheimer’s, cancer, and kidney, heart, and autoimmune diseases over the past 40 years in clinical practices. Along with many other polyphenolic actions (Figure 1), its versatility in multi-targeting disease mechanisms and maintaining cell homeostasis could therefore present polyphenols superior to any single drug in disease prevention.
Future directions
Wide variations of health benefits could exist with respect to polyphenol concentrations in different types of fruits and vegetables, the duration of polyphenol consumption, intestinal absorption and bioavailability, etc. Following the very limited intestinal absorption (<0.1%), question(s) remain whether polyphenolic catabolites/ metabolites by gut microbiota could likely exhibit/fuel beneficial or damaging health effects in addition to alteration of microbiota landscape [186-191]. Comprehensive and in-depth basic research and clinical trials could lay solid foundations in promoting polyphenols as phytotherapy, nutraceutics, or therapeutics. Further studies will afford polyphenols as essential components for human health in a new era of nutraceutics; several directions of future research certainly clarify and facilitate such advocate.
(1) The GI absorption, delivery, and catabolism could dictate the bioavailability of polyphenols. As a chemical, polyphenol’s “pharmacokinetics” and its possible intestinal precipitation remain unknown. Those lines of research including polyphenolic metabolism and catabolism could address the efficacy issues of polyphenols, ensuring disease prevention or intervention, if any. (2) Concerning diverse cellular effects by polyphenolic signaling, epigallocatechin 3-O-gallate, thus far, has shown to activate 67-kDa laminin receptor. Studies on the reception(s) could offer in-depth understanding and amplification/ramification of polyphenolicsignaling. (3) The effect of polyphenols on microbiota landscape remains in its infancy; thus far, enhanced GLP-1 secretion may be mediated by microbioa alteration [189]. In view of significant microbiota roles in metabolic disorders, further studies on such issues could elucidate diverse polyphenolic beneficial effects on metabolic diseases including obesity, diabetes, or CVD relevant to lipotoxicity, glucotoxicity, and/or other xenotoxicity. In this conjunction, do polyphenols directly affect metabolisms? (4) As prebiotics, polyphenols enhance the growth of healthy bacteria (Bifidobacterium, Lactobacillus, Akkermansia, and Klebsiella), while suppressing Clostridiales and Enterobacteriales. In addition to lowering CVD risks (e.g., diabetes, obesity, etc.), it will be interesting to learn the direct effects on CVD pathogenesis by such polyphenol-altered microbiota landscape. For instance, gut dysbiosis with low Bacteroidetes and high Firmicutes is associated with chronic hypertension in where there is also a decrease in acetate- and butyrate-producing bacteria and an increase in lactateproducing bacterial populations. Trimethylamine (TMA) formation from choline and carnitine/purine -rich diets by gut microbiota (e.g., Prevotella, Clostridium, Clostridiaceae, Preptostreptococcaceae, Lachnospira, etc.) could contribute to atherogenesis. Whether can polyphenols be of protection by suppressing those contributing microbiota for lowering such TMA formation? (5) Polyphenols are capable of enhancing GLP-1 [189] and adiponectin [169,170,182] secretion; it remains an open question whether polyphenols could globally modulate or interact with endocrine system(s) in relation to CVD. For instance, catecholamine impacts on ventricular hypertrophy, myocardial ischemia, and HF, while thyroid hormone could be assoicated with AF and protection from ischemic stress. Potential modulations on endocrine systems could further expand polyphenolic health benefits. (6) Further research on LPL upregulation and cholesterol catabolism could address polyphenol’s direct hypolipidemic actions. In view of cardioprotective HDL and atherogenic LDL, studies on the mechanism(s) by which polyphenols affect lipoprotein levels including Lp[a] could also offer insights into fighting CVD. The potentials to suppress PCSK-9/ Idol expression and functions could ascertain polyphenols’ actions on LDLR and clearance of plasma LDL-C as well as other TG-rich lipoproteins (e.g., VLDL, CM remnants, etc.). (7) MF polarization could dictate atherogenic progression/severity/manifestation as well as MI outcomes. MF polarization by polyphenols could extend the benefits to diverse inflammatory diseases including obesity, insulin resistance, metabolic symdrome, neurodegeneration, cancers, etc. (8) On the molecular biology fronts; green tea has shown to suppress DNA methylation. Food ingredients (e.g., vitamin coenzymes: flavin mononucleotide, thiamine pyrophosphate, B12) are known to participate in “RNA modulation” involving riboswitches [254]. It will be interesting to learn polyphenolic impacts on gene regulations/ modifications including epigenetic writer, readers, and erasers. Investigations on polyphenolic effects on microRNA (miR), if any, could also gain insights into enhanced protection or attenuated exacerbation/progression in CVD. Many cardiac miRs play either cardioprotective or pathogenic roles; they are coordinating in regulation and maintenance of cardiovascular functions [255]. For instance, miR- 21, 23a, 34a, 195, and 208a are positive, while miR- 133 is a negative regulator in cardiac hypertrophic processes. miR- 21 promotes cardiac hypertrophy and fibrosis mediated by ERK/ MAPK signaling. miR-34a modulates AT-II-induced myocardial hypertrophy. miR-33a is an atherogenic inducer, while miR-126 attenuates VCAM expression, lowering inflammation and leukocyte adherence to endothelial cells. miR-1 is a factor responsible for arrhythmias. MiR-499/24 protect from I/R injury, while miR-92a exacerbates it.
References
- Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. N Engl J Med. 2007; 356: 2388-2398.
- Yoon SS, Dillon CF, Illoh K, Carroll M. Trends in the prevalence of coronary heart disease in the U.S.: national health and nutrition examination survey, 2001–2012. Am J Prev Med. 2016; 51: 437-445.
- Eilat-Adar S, Sinai T, Yosefy C, Henkin Y. Nutritional recommendations for cardiovascular disease prevention. Nutrients. 2013; 5: 3646-383.
- Fernández-Velasco M, González-Ramos S, Boscá L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem J. 2014; 458: 187-193.
- B.W. Parks and A.J. Lusis. Macrophage accumulation in atherosclerosis. N Engl J Med.2013; 369: 2352-2353.
- Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost. 2017; 117: 7-18.
- Rodrigo R, Libuy M, Feliú F, Hasson D. Molecular basis of cardio protective effect of antioxidant vitamins in myocardial infarction. Biomed Res Int. 2013.
- Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016; 97: 153-161.
- You J, Wu J, Jiang G, Guo J, Wang S, et al. Olmesartan attenuates cardiac remodeling through DLL4/Notch1 pathway activation in pressure overload mice. J Cardiovasc Pharmacol. 2013; 61: 142-151.
- Matsushima S and Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015; 309: 1375-1389.
- Wende AR, O’Neill BT, Bugger H, Riehle C, Tuinei J, et al. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Mol Cell Biol. 2015; 35: 831-846.
- Hamamori Y and Schneider MD. HATs off to Hop: recruitment of a class I histone deacetylase incriminates a novel transcriptional pathway that opposes cardiac hypertrophy. J Clin Invest. 2003; 112: 824-826.
- Mishra S and Chatterjee S. Lactosylceramide promotes hypertrophy through ROS generation and activation of ERK1/2 in cardiomyocytes. Glycobiology. 2014; 24: 518-531.
- Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, et al. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002; 34: 233-240.
- Yang KC, Bonini MG, Dudley SC Jr. Mitochondria and arrhythmias. Free Radic Biol Med. 2014; 71: 351-361.
- Youn JY, Zhang J, Zhang Y, Chen H, Liu D, et al. Oxidative stress in atrial fibrillation: an emerging role of NADPH oxidase. J Mol Cell Cardiol. 2013; 62: 72-79.
- Schillinger KJ, Patel VV. Atrial fibrillation in the elderly: the potential contribution of reactive oxygen species. J Geriatr Cardiol. 2012; 9: 379-388.
- Joseph A Hill and Olson EN. Cardiac Plasticity. N Engl J Med. 2008; 358: 1370-1380.
- Thanassoulis G and Vasan RS. Genetic cardiovascular risk prediction- will we get there? Circulation. 2011; 122: 2323-2334.
- O’Donnell CJ and Nabel EG. Genomic Medicine: Genomics of cardiovascular disease. N Engl J Med. 2011; 365: 2098-2109.
- Nauseef WM. Biological Roles for the NOX Family NADPH Oxidases. J Biol Chem. 2008; 283: 16961-16965.
- Harrison R. Physiological roles of xanthine oxidoreductase. Drug Metab. Rev. 2004; 36: 363-375.
- Bryan N, Ahswin H, Smart N, Bayon Y, Wohlert S, Hunt JA. Reactive oxygen species (ROS) - a family of fate deciding molecules pivotal in constructive inflammation and wound healing. Eur Cell Mater. 2012; 24: 249-261.
- Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011; 208: 417-420.
- Bonizzi G, Piette J, Schoonbroodt S, Greimers R, Havard L, et al. Reactive oxygen intermediate-dependent NF-kappaB activation by interleukin-1beta requires 5-lipoxygenase or NADPH oxidase activity. Mol Cell Biol. 1999; 19: 1950-1960.
- Clark RA and Valente AJ. Nuclear factor kappa B activation by NADPH oxidases. Mech Ageing Dev. 2004; 125: 799-810.
- Wang X, Cui L, Joseph J, Jiang B, Pimental D, et al. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J Mol Cell Cardiol. 2012; 52: 753-760.
- Pollock DM and Pollock JS. Endothelin and oxidative stress in the vascular system. Curr Vasc Pharmacol. 2005; 3: 365-367.
- Wilkinson-Berka JL, Rana I, Armani R, Agrotis A. Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clin Sci. (Lond). 2013; 124: 597-615.
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2013; 35: 881-900.
- Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013; 25: 2185-2197.
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010; 464: 1357-13561.
- Stokes KY, Calahan L, Hamric CM, Russell JM, Granger DN. CD40/CD40L contributes to hypercholesterolemia-induced microvascular inflammation. Am J Physiol Heart Circ Physiol. 2009; 296: 689-697.
- ACC/AHA. The Guidelines battle on starting statins. N Engl J Med. 2014; 370: 1652-1658.
- Murad MH, Hazem A, Coto-Yglesias F, Dzyubak S, Gupta S, et al. The association of hypertriglyceridemia with cardiovascular events and pancreatitis: a systematic review and meta-analysis. BMC Endocr Disord. 2012; 5: 165-175.
- Durrington PN. Triglycerides are more important in atherosclerosis than epidemiology has suggested. Atherosclerosis. 1998; 141: 57-62.
- Gotto AM Jr. Triglyceride: the forgotten risk factor. Circulation. 1988; 97: 1027-1028.
- Aye MM, Kilpatrick ES, Aburima A, Wraith KS, Magwenzi S, et al. Acute hypertriglyceridemia induces platelet hyperactivity that is not attenuated by insulin in polycystic ovary syndrome. J Am Heart Assoc. 2014.
- Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007; 298: 299-308.
- Boffa MB and Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016; 57: 745-757.
- Sandoval YH, Atef ME, Levesque LO, Li Y, Anand-Srivastava MB. Endothelin-1 signaling in vascular physiology and pathophysiology. Curr Vasc Pharmacol. 2014; 12: 202-214.
- Mitchell JA, Ali F, Bailey L, Moreno L, Harrington LS. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp Physiol. 2008; 93: 141-147.
- Jin RC and Loscalzo J. Vascular nitric oxide: formation and function. J Blood Med. 2010; 1: 147-162.
- Ramanathan S, Mazzalupo S, Boitano S, Montfort WR. Thrombospondin-1 and angiotensin II inhibit soluble guanylyl cyclase through an increase in intracellular calcium concentration. Biochemistry. 2011; 50: 7787-7799.
- Johnson RJ, Feig DI, Nakagawa T, Gabriela Sanchez-Lozada L, Rodriguez- Iturbe B. Pathogenesis of essential hypertension: historical paradigms and modern insights. J Hypertens. 2008; 26: 381-391.
- Thomas RH. Hypercoagulability syndromes. Arch Intern Med. 2001; 161: 2433-2439.
- Jackson SP. Arterial thrombosis—insidious, unpredictable and deadly. Nat Medicine. 2011; 17: 1423-1436.
- Chu AJ. Blood coagulation as an intrinsic pathway for proinflammation. Infl Allergy Drug Targets. 201; 9: 32-44.
- Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflammation. 2011.
- Tappaelli C, Metternich R, Cook NS. Structure and function of thrombin receptors. Trends Pharmcol Sci. 1993; 14: 426-429.
- Soslau G, Class R, Morgan DA, Foster C, Lord ST, Marchese P, Ruggeri ZM. Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib. J Biol Chem. 2001; 276: 21173-21183.
- Adam F, Guillin MC, Jandrot-Perrus M. Glycoprotein Ib-mediated platelet activation. A signalling pathway triggered by thrombin. Eur J Biochem.2003; 270: 2959-2970.
- Vergnolle N, Derian CK, D’Andrea MR, Steinhoff M, Andrade-Gordon P. Characterization of thrombin-induced leukocyte rolling and adherence: a potential proinflammatory role for proteinase-activated receptor-4. J Immunol. 2002; 169: 1467-1473.
- Lin CC, Cooper DK, Dorling A. Coagulation dysregulation as a barrier to xenotransplantation in the primate. Transpl Immunol. 2009; 21: 75-80.
- Ware J. Thrombocytopathy and type 2B von Willebrand disease. J Clin Invest. 2013; 123: 5004-5006.
- Carter AM. Complement activation: An emerging player in the pathogenesis of cardiovascular disease. Scientifica (Cairo). 2012.
- Mandl-Weber S, Haslinger B, Sitter T. Thrombin up regulates production of plasminogen activator inhibitor type1in human peritoneal mesothelial cells. Perit Dial Int. 1999; 19: 319-324.
- Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-β1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008; 100: 976-983.
- Singh P, Peterson TE, Barber KR, Kuniyoshi FS, Jensen A, et al. Leptin upregulates the expression of plasminogen activator inhibitor-1 in human vascular endothelial cells. Biochem Biophys Res Commun. 2010; 392: 47- 52.
- Hao G-H, Niu X-L, Gao D-F, Wei J, Wang N-P. Agonists at PPAR-γ suppress angiotensin II-induced production of plasminogen activator inhibitor-1 and extracellular matrix in rat cardiac fibroblasts. Br J Pharmacol. 2008; 153: 1409-1419.
- Esterson YB, Kishore P, Koppaka S, Li W, Zhang K, et al. Fatty acid-induced production of plasminogen activator inhibitor-1 by adipose macrophages is greater in middle-aged versus younger adult participants. J Gerontol A Biol Sci Med Sci. 2012; 67: 1321-1328.
- Chen C, Nan B, Lin P, Yao Q. C-reactive protein increases plasminogen activator inhibitor-1 expression in human endothelial cells. Thromb Res. 2008; 122: 125-133.
- Bouma BN, Meijers JC. New insights into factors affecting clot stability: A role for thrombin activatable fibrinolysis inhibitor (TAFI; plasma procarboxypeptidase B, plasma procarboxypeptidase U, procarboxypeptidase R). Semin Hematol. 2004; 41: 13-19.
- van Tilburg NH, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood. 2000; 95: 2855-2859.
- Watanabe R, Wada H, Watanabe Y, Sakakura M, Nakasaki T, et al. Activity and antigen levels of thrombin-activatable fibrinolysis inhibitor in plasma of patients with disseminated intravascular coagulation. Thromb Res. 2001; 104: 1-6.
- Montaner J. Ribo M, Monasterio J, Molina CA, Alvarez-Sabin J. Thrombinactivable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke. 2003; 34: 1038-1040.
- Juhan-Vague I., Renucci JF, Grimaux M, Morange PE, Gouvernet J, Gourmelin Y, Alessi MC. Thrombin-activatable fibrinolysis inhibior antigen levels and cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2000; 20: 2156-2161.
- Guimaraes AH and Rijken DC. Thrombin activatable fibrinolysis inhibitor (TAFI) affects fibrinolysis in a plasminogen activator concentrationdependent manner. Study of seven plasminogen activators in an internal clot lysis model. Thromb Haemost. 2004; 91: 473-479.
- Schneider M, Brufatto N, Neill E, Nesheim M. Activated thrombin-activatable fibrinolysis inhibitor reduces the ability of high molecular weight fibrin degradation products to protect plasmin from antiplasmin. J Biol Chem. 2004; 279: 13340-13345.
- Belalcazar LM, Ballantyne CM, Lang W, Haffner SM, Rushing J, Schwenke DC, F. Pi-Sunyer X, Tracy RP, and the Look AHEAD (Action for Health in Diabetes) Research Group. Metabolic factors, adipose tissue and plasminogen activator inhibitor-1 levels in type 2 diabetes: Findings from the Look AHEAD Study. Arterioscler Thromb Vasc Biol. 2011; 31: 1689-1695.
- Foley JH, Nesheim ME, Rivard GE, Brummel-Ziedins KE. Thrombin activatable fibrinolysis inhibitor activation and bleeding in haemophilia A. Haemophilia . 2012; 18: 316-322.
- Adrogué HJ, Madias NE. Sodium and Potassium in the Pathogenesis of Hypertension. N Engl J Med. 2007; 356: 1966-1978.
- Mak IT, Landgraf KM, Chmielinska JJ, Weglicki WB. Angiotensin II promotes iron accumulation and depresses PGI and NO synthesis in endothelial cells: effects of losartan and propranolol analogs. Can J Physiol Pharmacol. 2012: 90: 1413-1418.
- Mishra PK, Tyagi N. Sen U, Joshua IG, Tyagi SC. Synergism in hyperhomocysteinemia and diabetes: role of PPAR gamma and tempol. Cardiovasc Diabetol. 2010.
- Cheng Z, Yang X, Wang H. Hyperhomocysteinemia and endothelial dysfunction. Curr Hypertens Rev. 2009; 5: 158-165.
- Schalinske KL, Smazal AL. Homocysteine Imbalance: a pathological metabolic marker. Adv Nutr. 2012; 3: 755-762.
- Chu AJ. Antagonisms by bioactive polyphenols against inflammation: a systematic view. Infl Allergy Drug Targets. 2014; 13: 34-64.
- Ross R. Atherosclerosis- an inflammatory disease. N Engl J Med. 1999; 340: 115-126.
- Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009; 27: 165-197.
- Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, Cotter PD. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes. 2012; 3: 186-202.
- Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2013; 249: 218- 238.
- Anfossi G, Russo I, Doronzo G, Pomero A, Trovati M. Adipocytokines in atherothrombosis: focus on platelets and vascular smooth muscle cells. Mediators Inflamm. 2010.
- Clària J, González-Périz A, López-Vicario C, Rius B, Titos E. New insights into the role of macrophages in adipose tissue inflammation and fatty liver disease: modulation by endogenous omega-3 fatty acid-derived lipid mediators. Front Immunol. 2011.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006; 444: 860-867.
- Wellen KE, Hotamisligil G.S. Inflammation, stress, and diabetes. J Clin Invest. 2005; 115: 1111-1119.
- Nathan C. Epidemic inflammation: pondering obesity. Mol Med. 2008; 14: 485-492.
- Ghesquie`re B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014; 511: 167-176.
- Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013; 34: 2436-243.
- Boden G. Obesity and free fatty acids (FFA). Endocrinol Metab Clin North Am. 2008; 37: 635-646.
- Funk SD, Yurdagul A, Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: Lessons from type 1 diabetes. Int J Vasc Med. 2012.
- Sprague JE, Arbeláez AM. Glucose counter regulatory responses to hypoglycemia. Pediatr Endocrinol Rev. 2011; 9: 463-475.
- Desouza CV, Bolli GB, Fonseca V. Hypoglycemia, diabetes, and cardiovascular events. Diabetes Care. 2010; 33: 1389-1394.
- Sherif EM, Elbarbary NS, Abd AAMM, Mohamed SF. Plasma thrombinactivatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: possible relation to diabetic microvascular complications. Blood Coagul Fibrinolysis. 2014; 25: 451-457.
- Vergeer M, Holleboom AG, Kastelein JJP, Kuivenhoven JA. The HDL hypothesis: does high-density lipoprotein protect from atherosclerosis? J Lipid Res. 2010; 51: 2058-2073.
- Casdorph HR. Hypercholesteremia-treatment with cholestyramine, a bile acid sequestering resin. Vasc Dis. 1967; 4: 305-308.
- Davignon J. Pleiotropic effects of pitavastatin. Br J Clin Pharmacol. 2012; 73: 518-535.
- Greenwood J, Mason JC. Statins and the vascular endothelial inflammatory response. Trends Immunol. 2007; 28: 88-98.
- Woodard J, Platanias LC. AMP-activated kinase (AMPK)-generated signals in malignant melanoma cell growth and survival. Biochem Biophys Res Commun. 2010; 398: 135-139.
- Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti-inflammatory effects of statins. Curr Opin Lipidol. 2011; 22: 165-170.
- Jougasaki M, Ichiki T, Takenoshita Y, Setoguchi M. Statins suppress interleukin-6-induced monocyte chemoattractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br J Pharmacol. 2010; 159: 1294-1303.
- Xing XQ, Duan S, Wu XW, Gan Y, Zhao SP, Chen P, Wu SJ. Atorvastatin reduces lipopolysaccharide-induced expression of C-reactive protein in human lung epithelial cells. Mol Med Rep. 2011; 4: 753-757.
- Townsend KP, Shytle DR, Bai Y, San N, Zeng J, et al. Lovastatin modulation of microglial activation via suppression of functional CD40 expression. J Neurosci Res. 2004; 78: 167-176.
- Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell.2013; 153: 112-125.
- Ehrenstein MR, Jury EC, Mauri C. Statins for atherosclerosis--as good as it gets? N Engl J Med. 2005; 352: 73-75.
- Meng X, Zhang K, Li J, Dong M, Yang J, An G, Qin W, Gao F, Zhang C, Zhang Y. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol Med. 2012; 18: 598-605.
- Goldfine AB. Statins: Is it really time to reassess benefits and risks? N Engl J Med. 2012; 366, 1752-1755.
- Recommendations for management of clinically significant drug-drug interactions with statins and select agents used in patients with cardiovascular disease: A scientific statement from the American Heart Association. 2016; 134: 468-495.
- Blom DJ, Hala T, Bolognese M, Lillestol MJ, Toth PD, et al. DESCARTES Investigators. A 52-Week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014; 370: 1809-1819.
- Lalanne F, Lambert G, Amar MJ, Chétiveaux M, Zaïr Y, et al. Wildtype PCSK9 inhibits LDL clearance but does not affect apoB-containing lipoprotein production in mouse and cultured cells. J Lipid Res. 2005; 46: 1312-1319.
- Chu AJ. Biochemical strategies to anticoagulation: A comparative overview. Curr Vasc Pharmacol. 2004; 2: 199-228.
- Baron TH, Kamath PS, McBane RD. Management of antithrombotic therapy in patients undergoing invasive procedures. N Engl J Med. 2013; 368: 2113- 2124.
- Schneider DJ. Anti-platelet therapy: glycoprotein IIb-IIIa antagonists. Br J Clin Pharmacol. 2011; 72: 672-682.
- Berny-Lang MA, Jakubowski JA, Sugidachi A, Barnard MR, Michelson AD, Frelinger AL. P2Y12 receptor blockade augments glycoprotein IIb-IIIa antagonist inhibition of platelet activation, aggregation, and procoagulant activity. J Am Heart Assoc. 2013.
- Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet Drugs: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012; 141: 89-119.
- Ghiassi-Nejadand Z, Friedman SL. Advances in anti-fibrotic therapy. Expert Rev Gastroenterol Hepatol. 2008; 2: 803-816.
- Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010; 38: 376-385.
- Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, et al. Tiplaxtinin, a novel orally efficacious inhibitor-1: Design, synthesis, and preclinical characterization. J Med Chem. 2004; 47: 3491-3494.
- Izuhara Y, Takahashi S, Nangaku M, Takizawa S, Ishida H, et al. Inhibition of plasminogen activator inhibitor-1. Its mechanism and effectiveness on coagulation and fibrosis. Arterioscler Thromb Vasc Biol. 2008; 28: 672-677.
- Willemse JL, Heylen E, Nesheim ME, Hendriks DF. Carboxypeptidase U (TAFIa): a new drug target for fibrinolytic therapy? J Thromb Haemost. 2009; 7: 1962-1971.
- Sacks FM, Campos H. Dietary therapy in hypertension. N Engl J Med. 2010; 362: 2102-2112.
- Hou J, Y. Kang J. Regression of pathological cardiac hypertrophy: Signaling pathways and therapeutic targets. Pharmacol Ther. 2012; 135: 337-354.
- Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. 2013; 24: 109-119.
- Chapman KE, Coutinho AE, Zhang Z, Kipari T, Savill JS, Seckl JR. Changing glucocorticoid action: 11β-Hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation. J Steroid Biochem Mol Biol. 2013; 137: 82-92.
- Serhan CN, Chiang N. Resolution phase lipid mediators of inflammation: agonists of resolution. Curr Opin Pharmacol. 2013; 13: 632-640.
- Prasain JK, Barnes S. Metabolism and bioavailability of flavonoids in chemoprevention: current analytical strategies and future prospectus. Mol Pharm. 2007; 4: 846-864.
- Liu Y, Hu M. Absorption and metabolism of flavonoids in the caco-2 cell culture model and a perused rat intestinal model. Drug Metab Dispos. 2002; 30: 370-377.
- Gundimeda U, McNeill TH, Fan TK, Deng R, Rayudu D, Chen Z, Cadenas E, Gopalakrishna R. Green tea catechins potentiate the neuritogenic action of brain-derived neurotrophic factor: role of 67-kDa laminin receptor and hydrogen peroxide. Biochem Biophys Res Commun. 2014; 445: 218-24.
- Mocanu MM, Ganea C, Georgescu L, Váradi T, Shrestha D, et al. Epigallocatechin 3-O-gallate induces 67 kDa laminin receptor-mediated cell death accompanied by downregulation of ErbB proteins and altered lipid raft clustering in mammary and epidermoid carcinoma cells. J Nat Prod. 2014; 77: 250-257.
- Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 2003; 111: 583-593.
- Prousek J. Fenton chemistry in biology and medicine. Pure Appl Chem. 2007; 79: 2325-2338.
- Deby-Dupont G, Mouithys-Mickalad A, Serteyn D, Lamy M, Deby C. Resveratrol and curcumin reduce the respiratory burst of Chlamydia-primed THP-1 cells. Biochem Biophys Res Commun. 2005; 333: 21-27.
- Chow SE, Hshu YC, Wang JS, Chen JK. Resveratrol attenuates oxLDLstimulated NADPH oxidase activity and protects endothelial cells from oxidative functional damages. J Appl Physiol. 2007; 102: 1520-1527.
- Petrônio MS, Zeraik ML, Fonseca LM, Ximenes VF. Apocynin: chemical and biophysical properties of a NADPH oxidase inhibitor. Molecules.2013; 18: 2821-2839.
- Huang XF, Li HQ, Shi L, Xue JY, Ruan BF, Zhu HL. Synthesis of resveratrol analogues, and evaluation of their cytotoxic and xanthine oxidase inhibitory activities. Chem Biodivers. 2008; 5: 636-642.
- Shen L, Ji HF. Insights into the inhibition of xanthine oxidase by curcumin. Bioorg Med Chem Lett. 2009; 19: 5990-5993.
- Aucamp J, Gaspar A, Hara Y, Apostolides Z. Inhibition of xanthine oxidase by catechins from tea (Camellia sinensis). Anticancer Res. 1997; 17: 4381- 4385.
- Schmidt AP, Böhmer AE, Antunes C, Schallenberger C, Porciúncula LO, Elisabetsky E, Lara DR, Souza DO. Anti-nociceptive properties of the xanthine oxidase inhibitor allopurinol in mice: role of A1 adenosine receptors. Br J Pharmacol. 2009; 156: 163-172.
- Nguyen MT, Nguyen NT. Xanthine oxidase inhibitors from Vietnamese Blumea balsamifera L. Phytother Res. 2012; 26: 1178-81.
- Bräunlich M, Slimestad R, Wangensteen H, Brede C, Malterud KE, Barsett H. Extracts, anthocyanins and procyanidins from Aronia melanocarpa as radical scavengers and enzyme inhibitors. Nutrients. 2013; 5: 663-678.
- Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer.2012; 12: 564-571.
- Chu AJ. Chapter 11: Phytochemicals in Disease Preventions and Interventions: A Current View. In: Introduction to Functional Food Science: Textbook (Volume 1); Martirosyan D.M. Ed.; Food Science Publishers. 2013.
- Manganiello V, Chung JH, Park SJ, Ahmad F, Philp A, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012; 148: 421-433.
- Mohar DS, Malik S. The Sirtuin system: The holy grail of resveratrol? J. Clin. Exp. Cardiolog. 2012; 3: 216.
- Jiang S, Wang W, Miner J, Fromm M. Cross regulation of sirtuin 1, AMPK, and PPARγ in conjugated linoleic acid treated adipocytes. PLoS One. 2012.
- Marchiani A, Rozzo C, Fadda A, Delogu G, Ruzza P. Curcumin and curcumin-like molecules: from spice to drugs. Curr Med Chem. 2013; 21: 204-222.
- Noorafshan A, Ashkani-Esfahani S. A review of therapeutic effects of curcumin. Curr Pharm Des. 2013; 19: 2032-2046.
- Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, Aggarwal BB. Multitargeting by curcumin as revealed by molecular interaction studies. Nat Prod Rep. 2011; 28: 1937-1955.
- Shehzad A, Lee YS. Molecular mechanisms of curcumin action: signal transduction. Biofactors. 2013; 39: 27-36.
- Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem. 2010; 10: 571-581.
- Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010; 285: 36387-36394.
- Johnson SM, Gulhati P, Arrieta I, Wang X, Uchida T, Gao T, Evers BM. Curcumin inhibits proliferation of colorectal carcinoma by modulating Akt/ mTOR signaling. Anticancer Res. 2009; 29: 3185-3190.
- Beevers CS, Chen L, Liu L, Luo Y, Webster NJ, Huang S. Curcumin disrupts the mammalian target of rapamycin-raptor complex. Cancer Res. 2009; 69: 1000-1008.
- Madan E, Prasad S, Roy P, George J, Shukla Y. Regulation of apoptosis by resveratrol through JAK/STAT and mitochondria mediated pathway in human epidermoid carcinoma A431 cells. Biochem Biophys Res Commun. 2008; 377: 1232-1237.
- Chung EY, Kim BH, Hong JT, Lee CK, Ahn B, Nam S, Han SB, Kim Y. Resveratrol down-regulates interferon-γ-inducible inflammatory genes in macrophages: molecular mechanism via decreased STAT-1 activation. J Nutr Biochem. 2011; 22: 902-909.
- Noh KT, Chae SH, Chun SH, Jung ID, Kang HK, Park YM. Resveratrol suppresses tumor progression via the regulation of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2013; 431: 348-353.
- Singh R, Ahmed S, Malemud CJ, Goldberg VM, Haqqi TM. Epigallocatechin- 3-gallate selectively inhibits interleukin-1beta-induced activation of mitogen activated protein kinase subgroup c-Jun N-terminal kinase in human osteoarthritis chondrocytes. J Orthop Res.2003; 21: 102-109.
- Zou MH, Hou XY, Shi CM, Nagata D, Walsh K, Cohen RA. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J Biol Chem. 2002; 277: 32552-32557.
- Manganiello V, Chung JH. Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, Kim MK, Beaven MA, Burgin AB. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012; 148: 421-433.
- Leiherer A, Mündlein A, Drexel H. Phytochemicals and their impact on adipose tissue inflammation and diabetes. Vascul Pharmacol. 2013; 58: 3-20.
- Shen M, Zhao L, Wu RX, Yue SQ, Pei JM. The vasorelaxing effect of resveratrol on abdominal aorta from rats and its underlying mechanisms. Vascul Pharmacol. 2013; 58: 64-70.
- Chung JH. Using PDE inhibitors to harness the benefits of calorie restriction: lessons from resveratrol. Aging (Albany NY). 2012; 4: 144-155.
- Marchiani A, Rozzo C, Fadda A, Delogu G, Ruzza P. Curcumin and curcumin-like molecules: from spice to drugs. Curr Med Chem. 2014; 21: 204-222.
- Noorafshan A, Ashkani-Esfahani S. A review of therapeutic effects of curcumin. Curr Pharm Des. 2013; 19: 2032-2046.
- Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, Aggarwal BB. Multitargeting by curcumin as revealed by molecular interaction studies. Nat Prod Rep. 2011; 28: 1937-1955.
- Bae JS. Role of high mobility group box 1 in inflammatory disease: focus on sepsis. Arch Pharm Res. 2012; 35: 1511-1523.
- Actis-Goretta L, Ottaviani JI, Fraga CG. Inhibition of angiotensin converting enzyme activity by flavanol-rich foods. J Agric Food Chem. 2006; 54: 229- 234.
- Reiter CE, Kim JA, Quon MJ. Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: roles for AMP-activated protein kinase, Akt, and FOXO1. Endocrinology. 2010; 151: 103-114.
- Wang A, Liu M, Liu X, Dong LQ, Glickman RD, Slaga TJ, Zhou Z, Liu F. Up-regulation of adiponectin by resveratrol: the essential roles of the Akt/ FOXO1 and AMP-activated protein kinase signaling pathways and DsbA-L. J Biol Chem. 2011; 286: 60-66.
- Tomé-Carneiro J, Gonzálvez M, Larrosa M, Yáñez-Gascón MJ, García- Almagro FJ, Ruiz-Ros JA, Tomás-Barberán FA, García-Conesa MT, Espín JC. Grape resveratrol increases serum adiponectin and downregulates inflammatory genes in peripheral blood mononuclear cells: a triple-blind, placebo-controlled, one-year clinical trial in patients with stable coronary artery disease. Cardiovasc Drugs Ther. 2013; 27: 37-48.
- Park CH, Tanaka T, Yokozawa T. Anti-diabetic action of 7-O-galloyl-Dsedoheptulose, a polyphenol from Corni Fructus, through ameliorating inflammation and inflammation-related oxidative stress in the pancreas of type 2 diabetics. Biol Pharm Bull. 2013; 36: 723-732.
- Kumar S, Narwal S, Kumar V, Prakash O. a-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn Rev. 2011; 5: 19- 29.
- Costa LG, Vitalone A, Cole TB, Furlong CE. Modulation of paraoxonase (PON1) activity. Biochem Pharmacol. 2005; 69: 541-550.
- Bijak M, Ziewiecki R, Saluk J, Ponczek M, Pawlaczyk I, Krotkiewski H, Wachowicz B, Nowak P. Thrombin inhibitory activity of some polyphenolic compounds. Med Chem Res. 2014; 23: 2324-2337.
- Kim DC, Ku SK, Bae JS. Anticoagulant activities of curcumin and its derivative. BMB Rep. 2012; 45: 221-226.
- Bijak M, Ponczek MB, Nowak P. Polyphenol compounds belonging to flavonoids inhibit activity of coagulation factor X. Int J Biol Macromol. 2014; 65: 129-135.
- Chen XQ, Wang XB, Guan RF, Tu J, Gong ZH, Zheng N, Yang JH, Zhang YY, Ying MM. Blood anticoagulation and antiplatelet activity of green tea (-)-epigallocatechin (EGC) in mice. Food Funct. 2013; 4: 1521-1525.
- Yamamoto E, Nishimura N, Okada K, Sekido C, Yamamichi S, Hasumi K. Inhibitors of autoactivation of plasma hyaluronan-binding protein (factor VII activating protease). Biol Pharm Bull. 2011; 34: 462-470.
- Milella RA, Antonacci D, Crupi P, Incampo F, Carrieri C, Semeraro N, Colucci M. Skin extracts from 2 Italian table grapes (Italia and Palieri) inhibit tissue factor expression by human blood mononuclear cells. J Food Sci. 2012; 77: 154-159.
- Bijak M, Bobrowski M, Borowiecka M, Podsedek A, Golanski J, Nowak P. Anticoagulant effect of polyphenols-rich extracts from black chokeberry and grape seeds. Fitoterapia . 2011; 82: 811-817.
- Olave NC, Grenett MH, Cadeiras M, Grenett HE, Higgins PJ. Upstream stimulatory factor-2 mediates quercetin-induced suppression of PAI-1 gene expression in human endothelial cells. J Cell Biochem. 2010; 111: 720-726.
- Cale JM, Li SH, Warnock M, Su EJ, North PR, Sanders KL, Puscau MM, Emal CD, Lawrence DA. Characterization of a novel class of polyphenolic inhibitors of plasminogen activator inhibitor-1. J Biol Chem. 2010; 285: 7892- 7902.
- Tomé-Carneiro J, Gonzálvez M, Larrosa M, Yáñez-Gascón MJ, García- Almagro FJ, Ruiz-Ros JA, García-Conesa MT, Tomás-Barberán FA, Espín JC. One-year consumption of a grape nutraceutical containing resveratrol improves the inflammatory and fibrinolytic status of patients in primary prevention of cardiovascular disease. Am J Cardiol. 2012; 110: 356-363.
- Sandra D, Radha M, Harishkumar M, Yuichi N, Sayuri O, Masugi M. Downregulation of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 by grape seed proanthocyanidin extract. Phytomedicine. 2010; 17: 42-46.
- Pan W, Chang MJ, Booyse FM, Grenett HE, Bradley KM, Wolkowicz PE, Shang Q, Tabengwa EM. Quercetin induced tissue-type plasminogen activator expression is mediated through Sp1 and p38 mitogen-activated protein kinase in human endothelial cells. J Thromb Haemost. 2008; 6: 976- 985.
- Abou-Agag LH, Aikens ML, Tabengwa EM, Benza RL, Shows SR, Grenett HE, Booyse FM. Polyphyenolics increase t-PA and u-PA gene transcription in cultured human endothelial cells. Alcohol Clin Exp Res. 2001; 25: 155- 162.
- Parkar SG, Stevenson DE, Skinner MA. The potential influence of fruit polyphenols on colonic microflora and human gut health. Int J Food Microbiology. 2008; 124: 295-298.
- Selma, MV, Espin JC, Tomas-Barberan FA. Interaction between phenolics and gut microbiota: role in human health. J Agri and Food Chemistry. 2009; 57: 6485-6501.
- Tzounis X, Vulevic J, Kuhnle GG, George T, Leonczak J, Gibson GR, Kwik- Uribe C, Spencer, JP. Flavanol monomer-induced changes to the human faecal microflora. Br J Nutr. 2008; 99: 782-792.
- Dao TM, Waget A, Klopp P, Serino M, Vachoux C, Pechere L, Drucker DJ, Champion S, Barthelemy S, Barra Y, Burcelin R, Seree E. Resveratrol increases glucose induced GLP-1 secretion in mice: a mechanism which contributes to the glycemic control. PLoS ONE. 2011; 6: 20700.
- Bialonska, D, Ramnani, P, Kasimsetty, SG, Muntha, KR, Gibson, GR, Ferreira, D. The influence of pomegranate by-product and punicalagins on selected groups of human intestinal microbiota. Int J Food Microbiology. 2010; 140: 175-182.
- Vodnar DC, Socaciu C. Green tea increases the survival yield of Bifidobacteria in simulated gastrointestinal environment and during refrigerated conditions. Chem Cent J. 2012; 6: 61.
- Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: Promising role for epigenetics. Biochem Pharmacol. 2010; 80: 1771-1792.
- Thakur VS, Gupta K, Gupta S. The Chemopreventive and chemotherapeutic potentials of tea polyphenols. Curr Pharm Biotechnol. 2012; 13: 191-199.
- Ussher JR, Drucker DJ. Cardiovascular actions of incretin-based therapies. Circ Res. 2014; 114: 1788-803.
- Ried K, Sullivan TR, Fakler P, Frank OR, Stocks NP. Effect of cocoa on blood pressure. Cochrane Database Syst Rev. 2012.
- Aviram M, Fuhrman B. Wine flavonoids protect against LDL oxidation and atherosclerosis. Ann N Y Acad Sci. 2002; 957: 146-161.
- Hernáez Á, Remaley AT, Farràs M, Fernández-Castillejo S, Subirana I and et al. Olive oil polyphenols decrease LDL concentrations and LDL atherogenicity in men in a randomized controlled trial. J Nutr. 2015; 145: 1692-1697.
- Wang GE, Li YF, Wu YP, Tsoi B, Zhang SJ, Cao LF, Kurihara H, He RR. Phloridzin improves lipoprotein lipase activity in stress-loaded mice via AMPK phosphorylation. Int J Food Sci Nutr. 2014; 65: 874-880.
- Yao N, He RR, Zeng XH, Huang XJ, Du TL, Cui JC, Hiroshi K. Hypotriglyceridemic effects of apple polyphenols extract via up-regulation of lipoprotein lipase in triton WR-1339-induced mice. Chin J Integr Med. 2014; 20: 31-35.
- Gutiérrez-Salmeán G, Ortiz-Vilchis P, Vacaseydel CM, Garduño-Siciliano L, Chamorro-Cevallos G, Meaney E, Villafaña S, Villarreal F, Ceballos G, Ramírez-Sánchez I. Effects of (-)-epicatechin on a diet-induced rat model of cardiometabolic risk factors. Eur J Pharmacol. 2014; 728: 24-30.
- Sikora J, Markowicz-Piasecka M, Broncel M, Mikiciuk-Olasik E. Extract of Aronia melanocarpa-modified hemostasis: in vitro studies. Eur J Nutr. 2014; 53: 1493-1502.
- Wallerath T, Deckert G, Ternes T, Anderson H, Li H, Witte K, Förstermann U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation. 2002; 106: 1652-1658.
- Speciale A, Chirafisi J, Saija A, Cimino F. Nutritional antioxidants and adaptive cell responses: an update. Curr Mol Med. 2011; 11: 770-789.
- Biasutto L, Mattarei A, Zoratti M. Resveratrol and health: the starting point. Chembiochem. 2012; 13: 1256-1259.
- Domitrovic R. The molecular basis for the pharmacological activity of anthocyans. Curr Med Chem. 2011; 18: 4454-4469.
- Thomasset S, Teller N, Cai H, Marko D, Berry DP, Steward WP, Gescher AJ. Do anthocyanins and anthocyanidins, cancer chemopreventive pigments in the diet, merit development as potential drugs? Cancer Chemother Pharmacol. 2009; 64: 201-211.
- Kanwar J, Taskeen M, Mohammad I, Huo C, Chan TH, Dou QP. Recent advances on tea polyphenols. Front Biosci. (Elite Ed). 2012; 4: 111-131.
- Whiting S, Derbyshire E, Tiwari BK. Capsaicinoids and capsinoids. A? A systematic review of the evidence. Appetite. 2012; 59: 341-348.
- Leiherer A, Mündlein A, Drexel H. Phytochemicals and their impact on adipose tissue inflammation and diabetes. Vascul Pharmacol. 2013; 58: 3-20.
- Siddiqui AM, Cui X, Wu R, Dong W, Zhou M, Hu M, Simms HH, Wang P. The anti-inflammatory effect of curcumin in an experimentalmodel of sepsis is mediated by up-regulation of peroxisome proliferator-activated receptor- . Crit Care Med. 2006; 34: 1874-1882.
- Tsuda S, Egawa T, Ma X, Oshima R, Kurogi E, Hayashi T. Coffee polyphenol caffeic acid but not chlorogenic acid increases 5’AMP-activated protein kinase and insulin-independent glucose transport in rat skeletal muscle. J Nutr Biochem. 2012; 23: 1403-1409.
- Akyol S, Ozturk G, Ginis Z, Armutcu F, Yigitoglu MR, Akyol O. In vivo and In vitro antineoplastic actions of caffeic ccid phenethyl ester (CAPE): therapeutic perspectives. Nutr Cancer. 2013; 65: 515-526.
- Landis-Piwowar K, Chen D, Foldes R, Chan TH, Dou QP. Novel epigallocatechin gallate analogs as potential anticancer agents: a patent review (2009 - present). Expert Opin Ther Pat. 2013; 23: 189-202.
- Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin- 3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011; 82: 1807-1821.
- Steinmann J, Buer J, Pietschmann T, Steinmann E. Anti-infective properties of epigallocatechin-3-gallate (EGCG), a component of green tea. Br J Pharmacol. 2013; 168: 1059-1073.
- Manganiello V, Chung JH, Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, Kim MK, Beaven MA, Burgin AB. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012; 148: 421-433.
- Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Physiol. 1998; 274: 692-699.
- Fryer LGD, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie G, Carling D. Activation of glucose transport by AMP-activated kinase via stimulation of nitric oxide synthesis. Diabetes. 2000; 49: 1978-1985.
- Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol. 1997; 273: 225.
- Kumar S, Narwal S, Kumar V, Prakash O. a-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn Rev. 2011; 5: 19- 29.
- Yousaf S, Butt MS, Suleria HA, Iqbal MJ. The role of green tea extract and powder in mitigating metabolic syndromes with special reference to hyperglycemia and hypercholesterolemia. Food Funct. 2014; 5: 545-556.
- Wang S, Moustaid-Moussa N, Chen L, Mo H, Shastri A, Su R, Bapat P, Kwun I, Shen CL. Novel insights of dietary polyphenols and obesity. J Nutr Biochem. 2014; 25: 1-18.
- Arias N, Macarulla MT, Aguirre L, Milton I, Portillo MP. The combination of resveratrol and quercetin enhances the individual effects of these molecules on triacylglycerol metabolism in white adipose tissue. Eur J Nutr. 2016; 55: 341-348.
- Saito M, Yoneshiro T. Capsinoids and related food ingredients activating brown fat thermogenesis and reducing body fat in humans. Curr Opin Lipidol. 2013; 24: 71-77.
- Higuchi M, Dusting GJ, Peshavariya H, Jiang F, Hsiao ST, Chan EC, Liu GS. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and Forkhead box O1 mediated upregulation of antioxidant enzymes. Stem Cells Dev. 2013; 22: 878-888.
- Malinowska J, Babicz K, Olas B, Stochmal A, Oleszek W. Aronia melanocarpa extract suppresses the biotoxicity of homocysteine and its metabolite on the hemostatic activity of fibrinogen and plasma. Nutrition. 2012; 28: 793-798.
- Domitrovic R. The molecular basis for the pharmacological activity of anthocyans. Curr Med Chem. 2011; 18: 4454-4469.
- Aboonabi A, Singh I. Chemopreventive role of anthocyanins in atherosclerosis via activation of Nrf2-ARE as an indicator and modulator of redox. Biomed Pharmacother. 2015; 72: 30-36.
- Matsushima S, Tsutsui H, Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc Med. 2014; 24: 202-205.
- Mokni M, Hamlaoui S, Karkouch I, Amri M, Marzouki L, Limam F, Aouani E. Resveratrol provides cardioprotection after ischemia/reperfusion injury via modulation of antioxidant enzyme activities. Iran J Pharm Res. 2013; 12: 867-875.
- Spinella F, Rosanò L, Di Castro V, Decandia S, Albini A, Nicotra MR, Natali PG, Bagnato A. Green tea polyphenol epigallocatechin-3-gallate inhibits the endothelin axis and downstream signaling pathways in ovarian carcinoma. Mol Cancer Ther. 2006; 5: 1483-1492.
- Li HL, Huang Y, Zhang CN, Liu G, Wei YS, Wang AB, Liu YQ, Hui RT, Wei C, Williams GM, Liu DP, Liang CC. Epigallocathechin-3 gallate inhibits cardiac hypertrophy through blocking reactive oxidative species-dependent and - independent signal pathways. Free Radic Biol Med. 2006; 40: 1756-1775.
- Thandapilly SJ, Louis XL, Yang T, Stringer DM, Yu L, Zhang S, Wigle J, Kardami E, Zahradka P, Taylor C, Anderson HD, Netticadan T. Resveratrol prevents norepinephrine induced hypertrophy in adult rat cardiomyocytes, by activating NO-AMPK pathway. Eur J Pharmacol. 2011; 668: 217-224.
- Nicholson SK, Tucker GA, Brameld JM. Effects of dietary polyphenols on gene expression in human vascular endothelial cells. Proc Nutr Soc. 2008; 67: 42-47.
- Dolinsky VW, Chakrabarti S, Pereira TJ, Oka T, Levasseur J, Beker D, Zordoky BN, Morton JS, Nagendran J, Lopaschuk GD, Davidge ST, Dyck JR. Resveratrol prevents hypertension and cardiac hypertrophy in hypertensive rats and mice. Biochim Biophys Acta. 2013; 1832: 1723-1733.
- Ahuja S, Kohli S, Krishnan S, Dogra D, Sharma D, Rani V. Curcumin: a potential therapeutic polyphenol, prevents noradrenaline-induced hypertrophy in rat cardiac myocytes. J Pharm Pharmacol. 2011; 63: 1604- 1612.
- Wongcharoen W, Phrommintikul A. The protective role of curcumin in cardiovascular diseases. Int J Cardiol. 2009; 133: 145-151.
- Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T, Hasegawa K. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008; 118: 868-878.
- Sheng R, Gu ZL, Xie ML. Epigallocatechin gallate, the major component of polyphenols in green tea, inhibits telomere attrition mediated cardiomyocyte apoptosis in cardiac hypertrophy. Int J Cardiol. 2013; 162: 199-209.
- Wu AZ, Loh SH, Cheng TH, Lu HH, Lin CI. Antiarrhythmic effects of (-)-epicatechin-3-gallate, a novel agonist in cultured neonatal rat ventricular myocytes. Biochem Pharmacol. 2013; 85: 69-80.
- Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016; 13: 167-179.
- Kim YS, Sayers TJ, Colburn NH, Milner JA, Young HA. Impact of dietary components on NK and Treg cell function for cancer prevention. Mol Carcinog. 2015; 54: 669-678.
- Casaer MP, Van den Berghr G. Nutrition in the acute phase of critical illness. N Engl J Med. 2014; 370: 1227-1236.
- Harris WS, Davidson MH, Schaefer EJ. Omega-3 fatty acids and coronary heart disease risk: clinical and mechanistic prospectives. Atherosclerosis. 2008; 197: 12-24.
- Jump DB, Depner CM, Tripathy S. Omega-3 fatty acid supplementation and cardiovascular disease: Thematic Review Series: New lipid and lipoproteint targets for the treatment of cardiometabolic diseases. J Lipid Res. 2012; 53: 2525-2545.
- De Caterina R. n–3 Fatty Acids in Cardiovascular Disease. N Engl J Med. 2011; 364: 2439-2450.
- Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014; 121: 91-119.
- Bao Y, Han J, Hu FB, Giovannucci EL, Stampfer MJ, Willett WC, Fuchs CS. Association of nut consumption with total and cause-specific mortality. N Engl J Med. 2013; 369: 2001-2011.
- Endo A, Kuroda M, Tanzawa K. Competitive inhibition of 3-hydroxy-3- methylglutaryl coenzyme A reductase 2 by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976; 72: 323- 326.
- Weissmann G. Aspirin. Sci Am. 1991; 264: 84-90.
- Fredman G, Serhan CN. Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem J. 2011; 437: 185-197.
- Hawley S, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Kirsty J. Mustard BE, Sakamoto KK, Steinberg GR, Hardie DG. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012; 336: 918-922.
- Shaw RJ, Cantley LC. Ancient sensor for ancient drug. Science. 2012; 336: 813-814.
- Johnson JE, Reyes FE, Polaski JT, Batey RT. B12 cofactors directly stabilize an RNA regulatory switch. Nature. 2012; 492: 133-137.
- Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013; 123: 11-18.