
Mini Review
J Cardiovasc Disord. 2015;2(1): 1010.
Novel Molecular Mechanism in Toll-Like Receptor-Related Inflammation in the Ischemic Brain
Munehisa Shimamura1,2*, Hironori Nakagami¹,Hideki Mochizuki² and Ryuichi Morishita³
¹Department of Health Development and Medicine, Osaka University Graduate School of Medicine, Japan
²Department of Neurology, Osaka University Graduate School of Medicine, Japan
³Department of Clinical Gene Therapy, Graduate School of Medicine, Osaka University, Japan
*Corresponding author: Munehisa Shimamura,Department of Health Development and Medicine, Center of Medical Innovation and Translational Research (6th floor, Room 0612B), Osaka University, 2-2 Yamada-oka,Suita, Osaka 565-0871, Japan
Received: December 16, 2014; Accepted: April 01, 2015; Published: April 02, 2015
Abstract
Although regulation of post-ischemic inflammation is an important strategy in treating ischemic stroke, most clinical trials have failed to show the benefit of modulation of microglia and macrophages; and inhibition of microglia, macrophages, and leukocyte recruitment in ischemic regions; and free radical scavenging except edaravone, which is the only free radical scavenger used in Japan, China. From the viewpoints, novel molecular and signaling mechanisms are required to develop new approaches in regulating post-ischemic inflammation. Examples of such approaches include regulation of inflammation through Toll-Like Receptors (TLRs) activation by Damage-Associated Molecular Pattern (DAMPs), which is released from the damaged neurons after cerebral ischemia. Recently, we have found that Receptor Activator of Nuclear Factor-Κb Ligand (RANKL) / receptor activator of nuclear factor-κB
(RANK) has been a novel signaling to be involved in TLR4-related inflammation and the regulation of RANKL/RANK signaling has a potential to be one of novel therapeutic approaches in ischemic brains. In this mini-review, we summarize recent findings on TLR-related inflammation in ischemic brain, especially focusing on RANKL/RANK signaling.
Keywords: Inflammation; Toll-Like Receptors; CD36; Peroxiredoxin; RANKL; RANK
Introduction
Regulation of post-ischemic inflammation is of interest to researchers because of its ability to prevent exacerbation of postischemic injury, including free radical scavenging; modulation of microglia and macrophages; and inhibition of microglia, macrophages, and leukocyte recruitment in ischemic regions. Edaravone is the only free radical scavenger accepted for use in Japan, China, and India; although it’s complete effectiveness needs to be confirmed further, in large, high-quality Trials [1]. However, other clinical trials on similar agents, including NXY-059, Tirilazad, Nicaraven, Ebselen, SUN-N8075 [2], minocycline [3], and uric acid [4], have failed to show efficacy. As a result, novel proteins and signaling processes have been explored to develop new approaches in treating post-ischemic inflammation. Among them, TLRs in activated macrophages and microglia have been extensively studied because molecular pattern molecules (DAMPs) released from damaged cells after cerebral ischemia strongly activate TLRs and leads to up regulation of proinflammatory gene expression through the transcription factor nuclear factor-kB [5]. One of strategies to regulate such TLRs-related inflammation is blocking DAMPs to activate the TLRs. A recent study has shown peroxiredoxin as a novel and powerful DAMP [6] although High Mobility Group Box 1 (HMGB1), beta-amyloid, and heat shock protein 70 (HSP 70) were previously proposed as being DAMPs. Extracellular peroxidrexin released from injured cells activates TLR2 and TLR4 in infiltrating immune cells, leading to production of cytotoxic cytokines, such as IL-23 and IL-17, leading to exacerbation of cerebral infarct. Because peroxiredoxin was released 12 h after ischemia, delayed treatment beyond the common therapeutic time window, i.e., 4.5 h after ischemia, was possible by blocking this signal with neutralizing antibody for peroxiredoxin [6]. Another strategy is to inhibit the TLRs signaling indirectly through inhibitory signaling for TLRs. As such a new signal regulating TLR signaling, we are focusing on a receptor activator of nuclear factor-κB ligand (RANKL) and a receptor activator of NF-κB (RANK) signaling in the ischemic brain [7].
RANKL/RANK Signaling in the Ischemic Brain
The idea of exploring RANKL/RANK signaling in the ischemic brain was led by epidemiological studies, which showed an association between severity and poor prognosis of ischemic stroke and high serum level of Osteoprotegerin (OPG), the decoy receptor for RANKL [8-10]. The function of OPG/RANKL/RANK signaling has been extensively studied in the field of bone metabolism, where RANKL induces osteoclast differentiation and activation through the receptor RANK. OPG inhibits this reaction as a decoy receptor for RANKL. However, these molecules are expressed in normal brain as well as immune cells such as macrophages, dendritic cells, and T cells. For example, RANKL/RANK signaling controls the thymocytemediated medulla formation and the formation of self-tolerance in T cells [11] as well as the number of regulatory T cells (Treg) [12]. In macrophages, RANKL directly regulates proinflammatory cytokine production [13]. OPG, RANKL, and RANK mRNA were expressed in normal brain; however, recent studies clarified that RANK was specifically expressed in astrocytes and neurons in the thermoregulatory center and that RANKL/RANK signaling contributes to fever and body temperature control [14]. Therefore, we examined the action of OPG/RANKL/RANK on the control of body temperature and inflammatory cytokines in the acute stage in the ischemic brain. We found that OPG/RANKL/RANK was expressed in activated microglia and that inflammatory cytokines and infarct volume were reduced, without affecting body temperature, when RANKL/RANK signaling was augmented using OPG-/- mice or mice treated with recombinant RANKL. In contrast, inhibition of RANKL/ RANK signaling, with neutralizing antibody for RANKL, resulted in increased inflammatory cytokines and infarct volume, indicating that RANKL/RANK signaling worked as an anti-inflammatory signal in microglia. In a mixed neuron/glia culture, stimulated by Lipopolysaccharide (LPS), which is a TLR4 ligand, addition of RANKL reduced neuronal death through the production of inflammatory cytokines. Because neuronal death could not be prevented by exogenous RANKL in glutamate, CoCl2, or H2O2, RANKL/RANK signaling does not act on neurons directly but on TLR signaling in microglia, and could potentially affect the outcome in ischemic brain [7] (Figure). In addition, we speculate that RANKL/RANK might be also a key signaling system in inducing immune tolerance in the chronic phase of the ischemic brain because a previous study showed that RANK was strongly expressed in dendritic cells in the periphery, where immune tolerance is induced by the activation of Tregs through RANKL/RANK signaling [12]. RANKL/RANK might be expressed in dendritic cells in the ischemic brain and contribute to immune tolerance, which is an important process for inhibition of autoantibody production in the injured brain [15]. Thus, activation of RANKL/RANK signaling seems a promising approach in the treatment of ischemic brain, but one of major problems is that effects of RANKL on causing osteoporosis [16] and vascular calcification [17]. Especially because just one i.p. injection of recombinant RANKL was shown to cause osteoporosis in mice [16], some modifications of RANKL itself or development of specific drug delivery systems into brain are needed to avoid osteoporosis or vascular calcification.
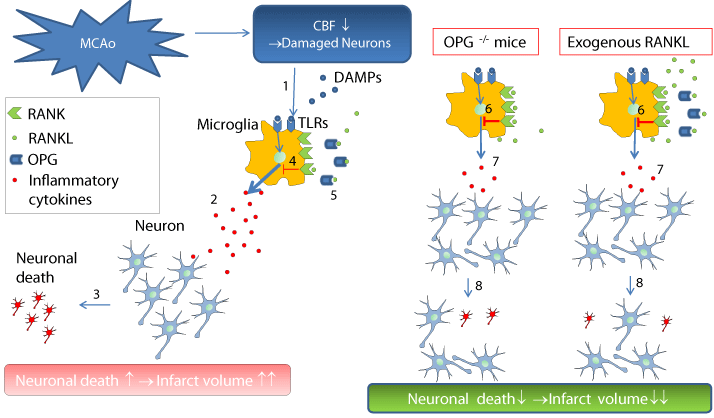
Figure 1: Schematic diagram of OPG/RANKL/RANK signaling in the
ischemic brain. DAMPs are released from damaged neurons after MCAo
[1] and increased TLR-induced inflammatory cytokines in the microglia [2],
this resulted in neuronal death and expansion of the infarct area [3]. RANKL
inhibits these responses through RANKL/RANK signaling [4], but OPG blocks
this inhibition [5] in WT mice. In OPG-/- mice or mice treated with exogenous
RANKL, the augmentation of RANKL/RANK signaling inhibits TLR-related
inflammatory cytokines [6,7] and prevents neuronal death [8]. Mcao: Middle
Cerebral Artery Occlusion; CBF: Cerebral Blood Flow; Damp: Damage-
Associated Molecular Pattern Molecules; Tlrs: Toll-Like Receptors; RANKL:
Receptor Activator Of Nuclear Factor-Κb Ligand; RANK: Nuclear Factor-Κb
Ligand (RANKL) - Receptor Activator Of NF- ?b: OPG: Osteoprotegerin
Conclusions
In the present review, we focused on molecules which affect TLRs signaling itself, but TLRs signaling also affects other signaling. For example, CD36 is a class-B scavenger receptor and its assembly with TLR2/1, but not TLR2/6, induces inflammatory cytokines in microglias and macrophages in brain and resulted in exacerbation of infarct volume [18]. Thus, TLRs-related inflammation is complexed and further study is necessary to clarify the molecular mechanisms. Although most anti-inflammatory agents targeting on classical signaling failed to show effects in clinical trials as mentioned in introduction, we believe that novel findings on post-ischemic inflammation might shed light on the development of therapeutic options in ischemic stroke.
References
- Feng S, Yang Q, Liu M, Li W, Yuan W, Zhang S, et al. Edaravone for acute ischaemic stroke. Cochrane Database Syst Rev. 2011; : CD007230.
- Kawakami M. Molecular Dissection of Cyclosporin A's Neuroprotective Effect Reveals Potential Therapeutics for Ischemic Brain Injury. Brain Sci. 2013; 3: 1325-1356.
- Kohler E, Prentice DA, Bates TR, Hankey GJ, Claxton A, van Heerden J, et al. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta-analysis. Stroke. 2013; 44: 2493-2499.
- Chamorro A, Amaro S, Castellanos M, Segura T, Arenillas J, Martí-Fábregas J, et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): a randomized, double-blind phase 2b/3 trial. Lancet Neurol. 2014; 13: 453-460.
- Iadecola 1, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011; 17: 796-808.
- Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012; 18: 911-917.
- Shimamura M, Nakagami H, Osako MK, Kurinami H, Koriyama H, Zhengda P, et al. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc Natl Acad Sci USA. 2014; 111: 8191-8196.
- Song TJ1, Kim J, Yang SH, Park JH, Lee HS, Nam CM, Lee OH. Association of plasma osteoprotegerin levels with stroke severity and functional outcome in acute ischaemic stroke patients. Biomarkers. 2012; 17: 738-744.
- Üstündag M, Orak M, Güloglu C, Tamam Y, Sayhan MB, Kale E. The role of serum osteoprotegerin and S-100 protein levels in patients with acute ischaemic stroke: determination of stroke subtype, severity and mortality. J Int Med Res. 2011; 39: 780-789.
- Jensen JK, Ueland T, Atar D, Gullestad L, Mickley H, Aukrust P, et al. Osteoprotegerin concentrations and prognosis in acute ischaemic stroke. J Intern Med. 2010; 267: 410-417.
- Ohigashi I, Nitta T, Lkhagvasuren E, Yasuda H, Takahama Y. Effects of RANKL on the thymic medulla. Eur J Immunol. 2011; 41: 1822-1827.
- Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med. 2006; 12: 1372-1379.
- Maruyama K, Takada Y, Ray N, Kishimoto Y, Penninger JM, Yasuda H, et al. Receptor activator of NF-kappa B ligand and osteoprotegerin regulate proinflammatory cytokine production in mice. J Immunol. 2006; 177: 3799-3805.
- Hanada R, Leibbrandt A, Hanada T, Kitaoka S, Furuyashiki T, Fujihara H, et al. Central control of fever and female body temperature by RANKL/RANK. Nature. 2009; 462: 505-509.
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011; 17: 796-808.
- Tomimori Y, Mori K, Koide M, Nakamichi Y, Ninomiya T, Udagawa N, et al. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J Bone Miner Res. 2009; 24: 1194-1205.
- Osako MK, Nakagami H, Shimamura M, Koriyama H, Nakagami F, Shimizu H, et al. Cross-talk of receptor activator of nuclear factor-?B ligand signaling with renin-angiotensin system in vascular calcification. Arterioscler Thromb Vasc Biol. 2013; 33: 1287–1296.
- Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, et al. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010; 41: 898-904.