
Research Article
J Cardiovasc Disord. 2015; 2(2): 1016.
Pathogenesis of Atherosclerosis in 2015 and Adventitial (Erzengin’s) Atherosclerocalcification
Erzengin F¹*, Bursuk E²
¹Department of Cardiology, Previous Dean, University of Istanbul, Istanbul Medical Faculty, Istanbul, Turkey
²Program of Biomedical Technologies, University of Istanbul, Vocational School of Technical Sciences, Istanbul, Turkey
*Corresponding author: Erzengin F, Department of Cardiology, Previous Dean, University of Istanbul, Istanbul Medical Faculty, Istanbul, Turkey
Received: June 18, 2015; Accepted: September 04, 2015; Published: September 08, 2015
Abstract
Formation of atherosclerotic and calcified plaque begins and grows up not only beneath the endothelium (intimae), but also in the adventitial subepithelium on the coronary arteries. MSCT is a unique non-invasive method for the detection of coronary atherosclerotic plaque morphology, and for the diagnosis of silent ischemia, lumen narrowing calcification of adventitia.
Keywords: Adventitial (Erzengin’s) atherosclerotic calcification; Formation of atherosclerotic calcification plaques on the adventitia; Multi slice computed tomographic angiography
Introduction
The term atherosclerosis refers to the thickened and hardened lesions, which have lipids and calcifications in the intimae and media of elastic and muscular arteries. To date, the primary cause of arterial atherosclerotic calcification has not yet been elucidated. It is generally accepted that atherosclerotic and calcified lesions first appear and develop within the innermost layer of the arteries (at the intimae). In 1995, the American Heart Association (AHA) described that the earliest lesions of atherosclerosis (fatty streaks or type III lesions) are present in the intimae of the aorta from childhood. Today, it is known that atherosclerosis begins as early as fetal life, especially in the fetuses of hypercholesterolemic mothers [1]. Formation and progression of atherosclerotic plaques and calcifications in all arterial beds of intimae have been well documented by many authors, such as V. Fuster and E. Falk [1-4], who subdivided the formation and progression of these plaques into several phases summarized below. As known, arterial calcification plays a major role in development of atherosclerosis and osteonecrosis protein is effective in the formation of this calcification [5]. Most lipids deposited in atherosclerotic lesions (atherosclerotic components) are derived from plasma Low-Density Lipoproteins (LDLs) that enter the vessel wall through injured or dysfunctional endothelium. Within the normal population, fatty streaks often appear in the aorta and coronary arteries starting from the ages of 5 to 10; these are considered as the initial/starting points of plaque development. Type I lesions consist of macrophage-derived foam cells that contain lipid droplets; Type II lesions contain macrophages and smooth muscle cells with extracellular lipid deposits; Type III lesions contain smooth muscle cells surrounded by extracellular connective tissue, fibrils, and lipid deposits; Type IV plaques consist of confluent cellular lesions with large amounts of extracellular lipid intermixed with fibrous tissue; and Type Va plaques possess an extracellular lipid core covered by a thin fibrous cap. Both phase III and IV plaques can evolve into phase V fibrotic plaques, characterized by type Vb or Vc lesions, with or without predominant calcification. Type VI lesions are occlusive thrombi overlying a superficial erosion of markedly stenotic and fibrocalcific phase V plaques [2-7]. In Phases II to V, vulnerable lipid-rich plaques are prone to disruption due to their high lipid content. Raised plaques appear later, generally by 20 years of age, and are present in areas such as the endothelium of proximal Left Anterior Descending (LAD) coronary artery, where fatty streaks are most prevalent in early life [3]. When fully opened, the total surfaces of all arteries in humans are approximately 800 square meters. Arteries produce more than 250 active substances. There are several types of plaque: Some are flat yellow dots or lines (fatty streaks), while others are raised above the surface as oval humps, which range in color from white to yellow (raised fibro lipid plaques) [1-7]. With early atherosclerotic lesions (fatty streaks or type III lesions) in the aortic root and coronary arteries, the lesion consists of lipid-laden monocyte-derived macrophage foam cells and a few T lymphocytes beneath an intact endothelium. It is generally accepted that only endothelial cells, monocyte-derived macrophages or foam cells, and a few T cells participate in the early inflammatory and immune responses that give rise to the early atherosclerotic lesions. These early atherosclerotic lesions (fatty streaks or type III lesions) represent a dynamic balance of the entry and exit of lipoproteins by endothelium injury, resulting in a predominance of lipoprotein exit and final scarring [1-7]. During the progression of the disease, the oxidative modified LDL (oxLDL), the inflammatory and immune response is accompanied by a fibro-proliferative response in which the vascular smooth muscle cells play dominant roles [1-12]. OxLDL has many proinflammatory properties, which explain the local up regulation of inducible endothelial cell adhesion molecules – even before lesion formation – in hypercholesterolemic atherosclerosis and plaque instability leading to atherothrombotic events [1-7]. OxLDL has proinflammatory and cytotoxic effects. It is recognized by the macrophage scavenger receptor, promoting intracellular lipid accumulation and foam cell formation. Such as the endothelial dysfunction or/activation (vascular cell adhesion molecule 1), monocyte adherence, injuring lipoprotein retention, oxLDL, inflammatory/immune response, macrophage scavenger foam cell formation, endothelial shear stress, turbulent flow, high blood pressure on the vessels’ walls and the role of Nitric Oxide (NO), prostacyclin, endothelin-1, acetylcholine, vascular Cell Adhesion Molecule-1 (CAM-1), Cell Adhesive Molecules (CAM or surface glycoproteins), Intercellular Adhesion Molecule-1 (ICAM-1) promote or enter though the injured or dysfunctional endothelium and subendothelial lipid accumulation and at the end lipid core formations occur. Monocytes (macrophages) adhere to the surface of endothelium (intimae) through specific molecules, such as the Monocyte Chemotactic Protein-1 (MCP-1) and Macrophage Colony-Stimulating Factor (M-CSF) [1]. Before or after their death, macrophage or foam cells can release various substances, including oxLDL and free radicals. During chronic minimal endothelial injury or dysfunction, plasma low-density proteins enter through the injured endothelium, leading to accumulation of lipids (oxLDL), while the macrophages produce atheroma masses or atheromatous component.
So this classical pathway or famous well known cascade of atherosclerosis brings about the formation of the atheroma in 1995 by AHA of atherosclerotic lesions [1-3]. This pathway of formation of atherosclerosis and calcifications almost universally were accepted in the intimae (under the endothelium). In spite of the fact that, there is no any doubt of this classical pathway or rule of the cascade of formation of atherosclerotic calcifications. Atherosclerosis is a focal disease in the intimae of large and medium sized systemic arteries. It has been well documented that, focal calcification in atherosclerotic plaques is very common in humans’ arteries and nicely shown that coronary calcification in adults is almost always atherosclerosis related intimae until today [9]. We have recently shown this with the MSCT method (previously 320 slices; currently 640 slices) using within a device which has a computerized magnifying glass. On the other hand making a comparison between MSCT, coronary angiography, perioperative findings and histopathological data which detected by light and electron microscopy and environmental SEM (By using a microtome, slice samples can be taken, across the walls of samples classical histopathological routine investigation and addition to chemical analysis can be taken by means of energy dispersive X-ray mobile monitoring the surface of specimen by SEM). We strongly emphasized that, the classical cascade of the atherosclerotic and calcified plaques formation appear and develop not only beneath the intimae (sub endothelium) or media, but also in adventitial epithelium on the coronary arteries. We have surprisingly determined that the adventitial side of the calcification develops much faster and more severely than that of the intimae and medial side on the arterial vessel walls.
MSCT of coronary angiography with 64 slice technologies was first described by Leschka S, et al [8]. Currently, we have performed our studies with MSCT at the beginning 64 slice and later on 320 slice with magnifying glass, which are very important tool for the non-invasive evaluation of coronary arterial pathologies. Moreover, the MSCT is an excellent and important technique for showing the location and size of plaque formation and calcification in the arteries [8-9]. It has been well documented in the last three decades that the cascade of atherosclerotic plaque formation begins from beneath the endothelial cells of the coronary arteries [1-23]. The media calcification of the coronary arteries was also described as Mönckeberg’s Sclerosis many years ago, and while rare in coronary arteries, it is frequent in other types of arteries (particularly in the arteries of the leg and aorta) [1,2,4,9,17,18]. All calcification of arteries are directly related with atherosclerotic burden or component [4,9,17,18]. Until today it has not written yet anything about our findings of adventitial atherosclerosis and calcifications [19-21]. In a two-year randomized study in 1997 on identical twin yeanlings, we determined that there were significant parallels between umbilical obesity and epicardial lipid mass. Moreover, we demonstrated that the epicardial lipid mass is statistically significantly associated with adventitial localization over the arteries [9].
We have recently shown with MSCT-320 and 640 that the formation of atherosclerotic and calcified plaques begin not only in the intimae, but, surprisingly, also under the adventitial epithelium of the coronary arteries. The adventitial calcifications develop more rapidly than the subendothelial (the intimae) calcified plaques. On the other hand, the medial calcifications develop far more frequently on the subendothelium of the aorta and its main branches, such as the carotid, vertebral, cerebral, renal, mesenteric, ilio-femoral, and peripheral arteries. Interestingly, the formation of calcified plaque begins more frequently beneath the subepithelium of the adventitia, and quickly develops towards to the lumen of arteries, following the same classical and accepted cascade on the coronary arteries. These calcifications mostly begin in the middle of the atheromatous component. Cholesterol (particularly oxLDL) and macrophage cells easily arrive to the adventitia vasa vasorum of the coronary arteries from blood and/or by diffusion from epicardial fat accumulation around the heart appears in three different types: a) Intracellular in the heart, b) Epicardial and c) Pericardial adipose tissue around the heart. Intracellular fat is the microscopic lipid accumulation within the cytoplasm cardiac myocytes, and can result from myocardial ischemia, cell damage and/or cell death. The epicardial fat tissue is located between the outer wall of the myocardium and the visceral layer of pericardium, while the pericardial fat exists anterior to the epicardial fat layer and therefore located between visceral and parietal pericardium. Due to the close anatomic relation between myocardium and the epicardial fat, the two tissues share the same microcirculation. Through the vasa vasorums, diffusion way or potential interactions through paracrine and vasocaine mechanism between epicardial fat and adventitia of coronary arteries or myocardium are strongly suggested [24,25] (Figure 1, Figures 3-10).
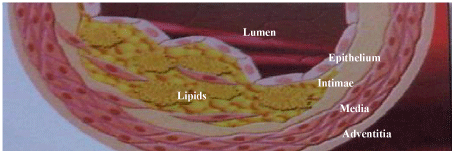
Figure 1: Classical pathway and accumulation of the lipids core in the intimae
have been shown in this figure.
Materials and Methods
Following animal studies in our advanced research center (after approval of the ethical committee), we examined coronary atherosclerotic calcifications by advanced technologies in 5750 patients at Istanbul Medical Faculty, and focused in particular on adventitial localization since 07.07.1997. Most risk factors apply to all atherosclerotic vascular diseases. In this context, few different consecutive cases (Figure1-10) were investigated by MSCT 320- 640 and conventional coronary angiography. The atherosclerotic plaques and calcifications appeared and developed on the adventitia inside or outside of the lumen of vessels on the coronary arteries (we identified the adventitial atherosclerosis for the first time in 2010) [19]. Our recent studies showed that in most patients (approximately 51%), the calcified plaques started just on the adventitia (Figures 8-9). In some cases, they can make a severe stenosis or occlusion of arterial lumen at the end. As we mentioned above, the adventitial atherosclerocalcification component can also grow outward of the artery (Figure 10). In comparison with IVUS, Stephan Achenbach et al found a sensitivity of 82% to detect coronary artery segments containing atherosclerotic plaque in patients without significant coronary artery stenosis [16]. MSCT might be very useful for the characterization of human coronary plaque morphology by noninvasively determining tissue density within the lesion. In addition, MSCT is a unique non-invasive method for the detection of coronary atherosclerotic plaque morphology, and for the diagnosis of silent ischemia and narrowing of lumen diameter by calcification of adventitia.
On Figure 1, the adventitial atherosclerotic calcifications (atheromatous components) on the femoral arteries of a) Guinea pig, b) Rabbit, c) Yeanling and d) Mouse have been shown. In the first patient, a normal coronary artery taken by MSCT was shown in Figure 2. In the third case (Figure 3), a soft plaque of atheroma on the right coronary artery of adventitia takes place. In the Figure 7 the atherosclerotic soft and vulnerable plaque (atheromatous component) starting from adventitia of the mid segmental part of the Left Anterior Descending (LAD) artery and multiple small adventitial calcifications were shown that just beneath the subepithelium of the adventitia on the (LAD). In Figures 3-10 have been shown from minimal to severe calcifications and some degree of atheromatous components were shown which started and were taken placed just beneath the epithelium of adventitia and grown towards to the coronary arterial lumen making some degree of obstructions on the Left Main, LAD and Circumflex (Cx) of coronary. MSCT-320 and 640 are very useful devices for the characterization of human coronary plaque morphology by determining tissue density within the lesion non-invasively.
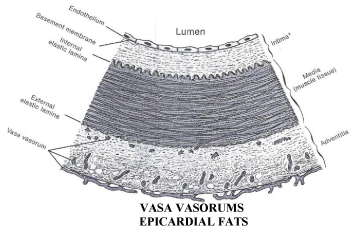
Figure 2: This figure shows epicardial fats tissue, vasa vasorums and tunicas
of an artery (endothelium, intimae, tunica elastica interna, media, tunica
elastica externa and adventitia).

Figure 3: This figure shows a normal artery (on the left), mild atherosclerosis
and remodelling of the artery ( in the middle) and lumen narrowing adventitial
(Erzengin’s) atherosclerosis (on the right).
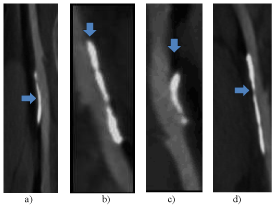
Figure 4: Adventitial atherosclerocalcifications (atheromatous components)
of on the femoral arteries of Guinea pig, b) Rabbit, c) Yeanling, d) Mouse. The
grey areas are soft plaque (lipids core), the white areas are calcified plaques.
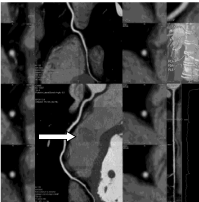
Figure 5: A normal human Right Coronary Artery (white arrow) is shown by
SCT with no atherosclerotic que or calcification.
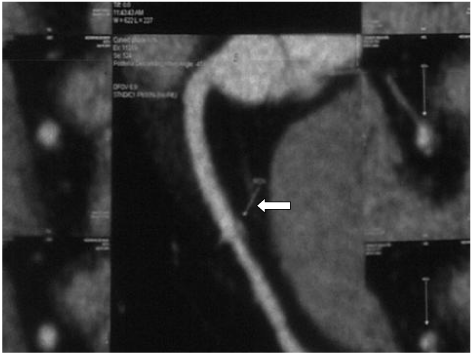
Figure 6: Adventitial soft plaque on the right coronary artery of a 48 years old
man (is shown white arrow).
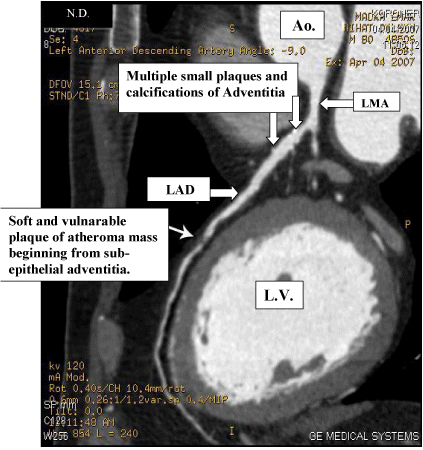
Figure 7: The adventitial atherosclerotic plaques with minimal calcifications
on the LAD artery without stenosis and a vulnerable soft plaque of
atheromatous component starting from subepithelial area of adventitia on
the mid LAD artery with significant stenosis. Ao: Aorta, LAD: Left Anterior
Descending, LMA: Left Main Artery, LV: Left Ventricle.
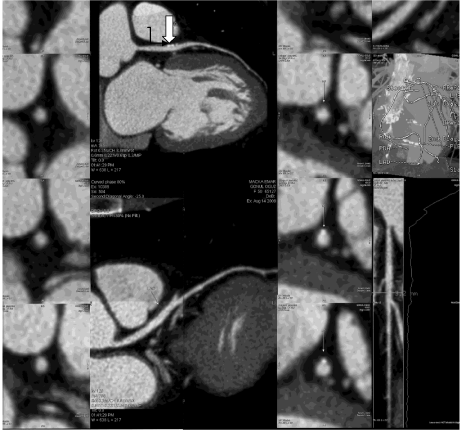
Figure 8: The atheroma mass; minimal calcifications surrounded by some
degree of (lipids) soft plaques (have been shown with black and white tiny
arrows) were taken place just below the adventitia of LAD artery. (In this
case, LAD and Cx arteries were emerging from two separated orifices as a
congenital coronary anomaly.)
Figure 9: Multi-size adventitial atherosclerocalcifications on the LAD and Cx
arteries.
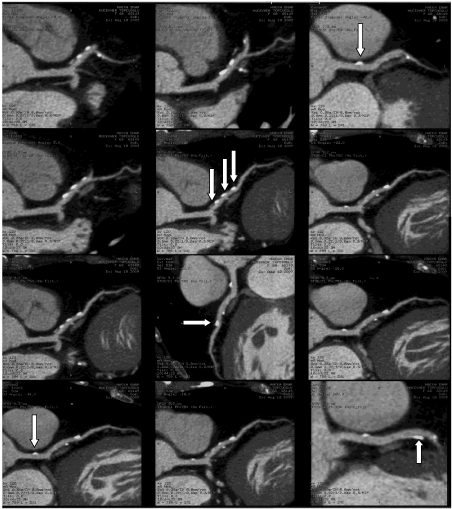
Figure 10: Consecutive many epithelial calcifications looks like beads on the
adventitia of the LAD artery and one calcified plaque on the adventitia of Left
Main Coronary Artery.
In our studies, a total of 5750 patients were investigated by MSCT, and most of them (73% of total patients) underwent invasive coronary angiography were performed to make a comparison between their results. At the end of this study, 793 patients had stent implantations, 323 patients underwent coronary bypass surgery and 4634 patients were treated medically. Also, MSCT is an important guide for planning elective treatment of lesions. The group of 5750 patients underwent MSCT and invasive coronary angiography, and more than 1000 had IVUS coronary arterial biopsy in the operating theater, with through cut coronary materials being sent immediately for light, electron and SEM microscopic investigations, which were performed to determine the localization of the atheromas, atheromatous components and calcifications.
Atherosclerotic calcification is one of the major causes of progressive degradation of the human arteries and target organs. It is important to note that the primary cause of arterial atherosclerotic calcification has not yet been elucidated. It progresses insidiously, and it damages to the human body remain unknown for a long time before symptoms and ischemia associated with the cardiac, cerebral, peripheral and other organs develop. Ischemia creates an imbalance between oxygen supply and demand of the tissue, and is usually related to atherosclerotic calcificated obstruction of the arteries, with more or less severe luminal stenosis, that may eventually be complicated by clot formation. Knowledge of its pathogenesis and of the early warning symptoms is imperative for interrupting the progression of this disease that not only has high mortality, but is also the cause of various disabling diseases.
It is currently known that atherogenesis is influenced by more than 250 factors. Atherosclerosis was defined for the first time by Osler in 1908 [9]. Osler mentioned that inflammation and infection were probably responsible for the etiology of atherosclerosis. Following this, the classical endothelial pathway of the atherosclerosis was accepted as the reason of atheroma formation in 1995 by AHA. Until today, it has been accepted that atherosclerotic calcification is located in the subendothelial area (intimae). Only Mönckeberg demonstrated several years ago the localization of atherosclerosis in the media among a limited group of patients [9]. According to the classical approach; it was known that this progressive process starts and grows up from intimae, which is inside the lumen of arteries, and also mostly progresses as time passes.
Results and Discussion
However, our team recently demonstrated the adventitial localization of atherosclerotic calcifications by using the Multi Slice Computed Tomography (MSCT) 64-320-640 with a magnifying glass on 5750 patients. Also, specimens of animals and patients who had bypass surgery had been investigated by pathology (light and SEM microscopy). Our conclusion was that the MSCT 320-640 is a very useful technique for the characterization of human coronary plaques morphology, for reason that the MSCT can non-invasively determine tissue density within lesions. In addition, as a unique and non-invasive method, the MSCT is used for the detection of coronary atherosclerotic plaque morphology (vulnerable plaques, in particularly) and calcification scores, also for the diagnosis of silent ischemia and asymptomatic myocardial infarctions caused by lumen narrowing due to calcification starting from the adventitia.
Thus, our surprising findings were that the formation of the atherosclerotic and calcified plaques begin and develop not only in the intimae or media (Mönckeberg’s Sclerosis), but also on the adventitia (Erzengin’s Arterial Atherosclerotic Calcifications) of the coronary arteries and/or all of the medium and large arteries [20]. We suggest that the formation of the Adventitial Atherosclerotic calcifications is caused by vasa-vasorums and/or diffusion from the adipose tissues around the pericardium.
As a result of our studies, these views were generated step-bystep as our knowledge has increased and become more refined during the last 5 years. It was the first time in the literature that the Adventitial Atherosclerotic Calcification was demonstrated by MSCT and proved pathologically in our research laboratories. In addition to this, coronary angiographies, IVUS, perioperative findings, histopathological data detected by light, electron microscopy and environmental SEM have been used for verifying the results.
For many years, atherosclerotic plaque formation has been accepted as a dynamic, progressive process and a very dangerous, fatal disease which can never be effectively prevented, stabilized or treated because of its dangerous progression. Unfortunately, atherosclerosis is still associated with high morbidity and mortality rates across the world.
According to our new clinical and laboratory findings; we believe that atherosclerotic and calcified plaques can be stabilized and fully treated with only drug therapy, regardless of where their localization is on the arteries. This means that, in addition to the current treatment, our polypill combination can totally cure the atherosclerosis and prevent plaque formation permanently. So, we have proved that this insidious and potentially fatal process is regressive, preventable and totally curable using only a combined drug therapy.
Conclusion
According to our study, new approach to the pathogenesis of atherosclerosis has been described in our studies, so formation of atherosclerotic and calcified plaque begins and grows up not only beneath the endothelium (intimae), but also in the adventitia on the coronary arteries.
References
- Fuster V. Epidemic of cardiovascular disease and stroke: the three main challenges. Presented at the 71st scientific sessions of the American Heart Association. Dallas, Texas. Circulation. 1999; 99: 1132-1137.
- Fuster V. Atherosclerosis, thrombosis, and vascular biology. Cecil Textbook of Medicine. Lee Goldman, Dennis Ausiello, editors. 22nd edn. Saunders, Philadelphia, Philadelphia. 2004; 383-400.
- Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998; 339: 1349-1357.
- Falk E, FusterV. Atherogenesis and its determinants. Hurst’s The Heart. Valentin Fuster, Wayne Alexander R, Robert A. O’Rourke, Robert Roberts, Spencer B. King III, Hein JJ Wellens, editors. 10th edn. McGraw-Hill, New York. 2001; 1: 1065-1093.
- Scicchitano P, Marzullo A, Ciccone MM. The role of intimal arterial calcification in the context of atherosclerotic plaque stability. Cytology and Histology. 2014; 5: 1000e111.
- Ross R. The biology of atherosclerosis. Topol EJ, editors. In: Comprehensive Cardiovascular Medicine. Philadelphia: lippincott-Raven, 1998: 13.
- Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999; 85: 199-207.
- Leschka S, Alkadhi H, Plass A, Desbiolles L, Grünenfelder J, Marincek B, et al. Accuracy of MSCT coronary angiography with 64-slice technology: first experience. Eur Heart J. 2005; 26: 1482-1487.
- Erzengin F, Büyüköztürk K. Cardiac Imaging in Textbook of Internal Medicine, Kemalettin Büyüköztürk, Faruk Erzengin, editors. Nobel, Istanbul. 2007; 1623-1644.
- Westendorp RG, Assendelft WJ, Elzen WP, Cessie S, Gussekloo J. Use of Framingham risk and new biomarkers to predict cardiovascular mortality in older people: Population base observational cohort study. BMJ. 2009; 338: a3083.
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adult (Adult Treatment Panel III) final report. Circulation. 2002; 106: 3143-3421.
- Conroy RM, Pyorala K, Fitzgerald AP, Sans S, Menoti A, De Backer G. Estimation of ten year risk of fatal cardiovascular disease in Europe: The SCORE Project. European Heart Journal. 2003; 290: 898-904.
- Hendel RC, Patel MR, Kramer CM, Poon M. ACC/ACR/SCMR/ASNC /NASCI/ SCAI/SIR Apropriateness Criteria for Cardiac Computed Tomography and Cardiac Magnetic Resonance Imaging. Journal of the American College of Cardiology. 2006; 48: 1-23.
- Achenbach S, Daniel WG. Computed Tomography of the Heart. Braunwald’s Heart Disease, 7th edition, Elsevier Saunders, Pensilvania, USA. 2005; 285-287.
- Higgins CB, de Roos A. MRI and CT of the Cardiovascular system. Lipincor &Williams and Wilkins,USA and the Netherlands, 2006.
- Achenbach S, Moselewski F, Ropers D, Ferencik M, Hoffmann U, MacNeill B, et al. Detection of calcified and noncalcified coronary atherosclerotic plaque by contrast-enhanced, submillimeter multidetector spiral computed tomography: a segment-based comparison with intravscular ultrasound. Circulation. 2004; 109: 14-17.
- Sangiorgy G, Rumberg JA, Severson A, et al. Arterial calcificationand not lumen stenosis is highly correlated with atherosclerotic plaqueburden in humans: A histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol. 1998; 31: 126-133.
- Mintz GS, Pichard AD, Popma JJ, Kent KM, Satler LF, Bucher TA, Leon MB. Determinants and correlates of target lesion calcium in coronary artery disease: a clinical, angiographic and intravascular ultrasound study. J Am Coll Cardiol. 1997; 29: 268-274.
- Mintz GS, Pichard AD, Popma JJ, Kent KM, Satler LF, Bucher TA, Leon MB. Determinants and correlates of target lesion calcium in coronary artery disease: a clinical, angiographic and intravascular ultrasound study. J Am Coll Cardiol. 1997; 29: 268-274.
- grow up not only beneath the endothelium, but also on the adventitia. 78th European Atherosclerosis Society Congress (78th EAS Congress) Hamburg, Germany. 2010; 20-23.
- Erzengin F. Adventitial (Erzengin’s) Atherosclrero-calcification: Surprise Location. Atherosclerotic and
- calcified plaques begin and grow up not only beneath the endothelium, but also on the adventitia. THE 3th WORLD MEDICINE CONGRES In abstract book. NALCHIK-RUSSIA. 2010; 20-23.
- Erzengin F. Adventitial arherosclerocalcifications: A new surprise location, In Abstract book, 7th Congress of update in Cardiology and Cardiovascular Surgery in association with TCT Mediterranean 24-27th March. ANTALYA-TURKEY. 2011.
- Bissen EA. Plaque Adventitia: Service Hatch or Battleground? 79th EAS Congress Gothenburg, Sweden, June 26-29, 2011.
- Efe D, Aygün F. Assessment of the relationship between non-alcoholic fatty liver disease and CAD using MSCT. Arq Bras Cardiol. 2014; 102: 10-18.
- Lee SY, Chang HJ, Sung J, Kim KJ, Shin S, Cho IJ, et al. The impact of obesity on subclinical coronary atherosclerosis according to the risk of cardiovascular disease. Obesity (Silver Spring). 2014; 22: 1762-1768.
- Iacobellis G, Ribaudo MC, Zappaterreno A, Iannucci CV, Leonetti F. Relation between epicardial adipose tissue and left ventricular mass. Am J Cardiol. 2004; 94: 1084-1087.