
Review Article
J Cardiovasc Disord. 2016; 3(1): 1021.
Mouse Models of Coronary Artery Atherosclerosis
Gonzalez L1,2, Yu P1,2 and Trigatti BL1,2*
1Department of Biochemistry and Biomedical Sciences, McMaster University, Canada
2Thrombosis and Atherosclerosis Research Institute, McMaster University, Canada
*Corresponding author: Bernardo Trigatti, Department of Biochemistry and Biomedical Sciences, and Thrombosis and Atherosclerosis Research Institute, McMaster University, Canada
Received: July 25, 2015; Accepted: January 18, 2016; Published: January 20, 2016
Abstract
Atherosclerosis is a chronic disease affecting large- and medium-sized arteries and it is the main underlying cause of cardiovascular diseases. Animal models, particularly mouse models, represent powerful tools to uncover disease mechanisms. Through a combination of genetic and diet manipulation, several mouse models for atherosclerosis research have been developed, with the apolipoprotein E and low-density lipoprotein receptor models being the most widely used. However, these mouse models remain relatively resistant to atherosclerosis development in coronary arteries and development of atherosclerosis related myocardial infarction, key features of human atherosclerotic disease. The discovery that the scavenger receptor class B type 1 acted as a high affinity high-density lipoprotein receptor and the inactivation of its gene in mice allowed for the generation of new models exhibiting either spontaneous or diet-induced occlusive coronary artery atherosclerosis and myocardial infarction. This review will discuss mouse models of coronary heart disease, highlighting their characteristics and focusing on those dependent on scavenger receptor class B type 1 deficiency.
Keywords: Atherosclerosis; Mouse; Coronary arteries
Abbreviations
APO: Apolipoprotein; APOBEC-1: apoB mRNA Editing Catalytic Polypeptide-1; CA: Coronary Artery; CAD: Coronary Artery Disease; CVD: Cardiovascular Disease; DKO: Double Knockout; eNOS: Endothelial Nitric Oxide Synthase; HDL: High-Density Lipoprotein; HDL-C: HDL Cholesterol; HL: Hepatic Lipase; ICAM: Intercellular Adhesion Molecule; IDL: Intermediate-Density Lipoprotein; KO: Knockout; LDL: Low-Density Lipoprotein; LDLR: LDL Receptor; LRP: LDLR-Related Protein; PDZ: Postsynaptic Density Protein-95, Drosophila disc-large protein, Zonula occludens protein 1; PDZK1, PDZ Containing 1; Plg: Plasminogen; SMC: Smooth Muscle Cells; SR-B1: Scavenger Receptor Class B Type 1; Tg: Transgenic; tKO: triple Knockout; TNF: Tumor Necrosis Factor; uPA: urokinase Plasminogen Activator; VCAM: Vascular Adhesion Molecule; VLDL: Very Low-Density Lipoprotein
Introduction
Despite the great advances in cardiovascular research in the past decades, Cardiovascular Disease (CVD) remains the main cause of death in western societies [1,2]. In the year of 2012, about 15.5 million US adults had CVD, among which 7.6 million were diagnosed with myocardial infarction [3]. It has been estimated that in the US only, 40.5 % of the population will present some form of CVD by 2030 [4]. Alongside the increase in the prevalence of CVD worldwide, the cost associated with the treatment of these conditions also increases dramatically. The medical cost associated with the treatment of Coronary Artery Disease (CAD) in the US is projected to increase by approximately 200 % in a span of 20 years [5]. Hence, efforts are concentrated on understanding and characterizing the several factors that govern the initiation and progression of CAD. Atherosclerosis, the main underlying pathology in CAD, is a chronic inflammatory disease of the artery wall that affects large- and medium-sized arteries [1]. The presence of early-stage atherosclerotic lesions (intima-media thickening) can be detected in young adults (adolescence), and the bulk of the disease develops during adulthood [6,7].
Atherosclerosis is characterized by the accumulation of modified lipoproteins in the intimal layer of the vessel wall, which triggers the expression of adhesion molecules on the surface of the endothelium (Figure 1) [8]. In addition to the expression of adhesion molecules, the endothelial cells can also release chemokines triggering the recruitment, from circulation, of immune cells, mainly monocytes that will differentiate into macrophages [9]. These mononuclear phagocytes can now ingest the aggregated modified lipoproteins becoming foam cells. Lipid-laden macrophage foam cells may further contribute to the pro-inflammatory environment in the vessel wall by releasing cytokines like interleukin-1β and Tumor Necrosis Factor (TNF)-a [2,10]. The formation of the atherosclerotic plaque also involves the recruitment and activation of Smooth Muscle Cells (SMC) from the tunica media into the intima. Recruited SMC secrete extracellular matrix proteins, like collagen and elastin, and accumulate on top of the lesion generating the fibrous cap [11]. Foam cells, cell debris and extracellular lipid accumulate underneath the fibrous cap generating the pro-thrombotic necrotic core. The rupture of the fibrous cap, and exposure of the contents of the lipidrich necrotic core to blood, trigger thrombus formation leading to myocardial infarction/stroke [12].
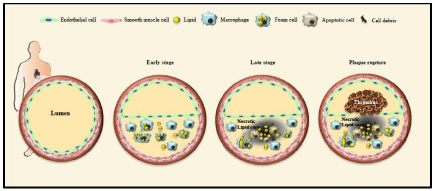
Figure 1: Atherosclerotic plaque initiation and progression.
At early stages, vessel wall inflammation and accumulation of modified lipoproteins trigger the recruitment of monocytes into the intima and their differentiation into
macrophages. Macrophages will engulf the modified lipoproteins becoming foam cells. Foam cells can release cytokines and chemokines further amplifying the
response. At late stages, accumulation of foam cells, apoptotic cells, cell debris and extracellular lipid creates the necrotic lipid core. Plaques with larger necrotic
cores are unstable and contain thinner fibrous caps, which facilitates plaque rupture and exposure of plaque contents to circulation, triggering blood coagulation
and thrombus formation.
Intense research has been dedicated to elucidating the mechanisms behind atherosclerotic plaque formation in hopes of finding new targets for disease treatment and prevention. Early studies in the atherosclerosis field employed rabbits as the main animal model system, with some studies using swine and non-human primates [13-15]. The mouse has come to dominate the research field as the animal model of choice for atherosclerosis studies, mainly due to the ease of genetic manipulation, facilitating analysis of mechanisms of atherosclerotic disease development. This review will discuss the main features of the most commonly used mouse models for the study of atherosclerosis with a special emphasis in models of CAD (Table 1).
![]()
Model
Characteristics
High fat or cholesterol diet required
Time
frame
Reference
apoE/LDLR dKO
Significant hypercholesterolemia CA atherosclerosis after long term high fat diet feeding. MI induced by stress in an endothelin-dependent fashion.
Yes 21% fat, 0.15 % cholesterol
20-24 weeks feeding
[52]
SR-B1/apoE dKO
Mice are hypercholesterolemic and spontaneously develop lesions in the aortic sinus and CA’s. Mice develop MI, cardiac dysfunction. No CA plaques are observed at 3 weeks of age. CA plaques and MI are observed by 5 weeks of age. Mice do not survive beyond 8-9 weeks of age.
No
2-5 weeks
[70,71,79,81]
SR-B1 KO /apoE-hypomorphic
Mice develop diet-induced hypercholesterolemia and aortic root and CA atherosclerosis. Occlusive CA disease leads to myocardial infarction, cardiac dysfunction and premature death.
Yes Paigen diet
3.5 weeks
feeding
[84]
SR-B1/LDLR dKO
Lesions develop on the aortic root in high fat diet-fed mice. The severity of the disease is dependent on the cholesterol and cholate content of the diet. Mice rapidly develop high levels of CA atherosclerosis and reduced survival due to myocardial infarction. Lesions also presented diet-dependent platelet accumulation.
Yes
Paigen diet
Cholate-free
Paigen diet
2 % cholesterol
3.5 weeks
feeding
6 weeks
feeding
9 weeks
feeding
[90,92]
PDZK1/apoE dKO
Mice present significant reduction on SR-B1 levels in liver and intestine. Lesions develop in a diet-dependent fashion. Mice fed a Paigen diet presented significant atherosclerosis in both the aortic sinus and CAs. No effect on cardiomegaly or lifespan.
Yes Paigen diet
12 weeks
feeding
[102,106]
eNOS/apoE dKO
Mice develop high fat diet induced CA atherosclerosis, aortic aneurysm and myocardial ischemia with no effect on survival.
Yes� 21% fat, 0.15 % cholesterol
16 weeks
feeding
[110]
AKT1/apoE dKO
Atherosclerosis develops in aortic sinus, aorta and CA after high cholesterol diet feeding. Development of the disease is attributed to an increase in apoptosis and macrophage infiltration. Myocardial infarction and reduced survival are also features of the model.
Yes 1.25 % cholesterol
14 weeks feeding
[113]
uPA-Tg/apoE KO
Mice exhibit diet induced CA atherosclerosis, myocardial fibrosis and reduced survival. Myocardial fibrosis may be independent of CA disease. Phenotype is dependent on the presence of plasminogen.
Yes 21% fat, 0.15 % cholesterol
10 weeks feeding
[117,119]
Table 1: Mouse Models of Coronary Artery Atherosclerosis.
Mouse models of atherosclerosis
Despite the attractive characteristics of the mouse as a model of atherosclerosis, no inbred strain is known to develop atherosclerotic plaques spontaneously. Mice carry most of their plasma cholesterol in High-Density Lipoproteins (HDL), while in humans the main cholesterol carrier is the Low-Density Lipoprotein (LDL) class [16]. This difference between species might contribute to the protection seen in the mouse against atherosclerosis, since HDL is considered to be protective against CVD, while high LDL cholesterol is considered as a risk factor [17]. In 1985, Paigen and colleagues reported the results of feeding different inbred strains of mice a high fat, high cholesterol diet, containing cholate, in an effort to identify atherosclerosis susceptibility differences between strains [18]. The results of the study established that C57BL/6 mice were the most sensitive, whereas C3H mice were the most resistant to diet-induced atherosclerosis development. A gender difference has also been detected under similar conditions, with female mice being more susceptible to plaque development [19]. Yet, the atherosclerotic lesions in C57BL/6 mice on the “Paigen diet” arose only in the aortic root and the most proximal aorta and displayed features of early stages of plaque development [20,21]. Studies by Liao et al. suggested that the genetic differences in atherosclerosis susceptibility seen in the inbred strains of mice might be a consequence of differences in their inflammatory responses, suggesting the importance of inflammation in atherosclerosis development [22]. The advent of gene targeting approaches in mice led to the generation of two widely used mouse models for the study of atherosclerosis: the apolipoprotein (apo) E Knockout (KO) and LDL receptor (LDLR) KO mouse models.
ApoE KO mouse model
ApoE is a protein present on the surface of Very Low-Density Lipoproteins (VLDL), HDL and chylomicrons. A major role of apoE is to mediate binding of apoE-containing lipoproteins to the LDLR, serving as a ligand for clearance of these lipoproteins by the liver [23]. In 1992, two groups independently reported the generation of and the characteristics of atherosclerosis development in apoE KO mice [24-26]. Since then, several studies have contributed to evidence of its pleiotropic role on atherosclerosis development.
The absence of apoE in mice results in a phenotype resembling human type III hyperlipoproteinemia, with severe hypercholesterolemia due to accumulation of lipoprotein remnants, even on a normal chow diet [27]. Reports have shown that on a normal chow diet, apoE KO mice spontaneously develop lesions in the aortic sinus and aortic branches with foam cells appearing as early as 8 weeks of age [28]. High-fat diet feeding accelerated lesion development and led to more advanced lesions similar in complexity to plaques found in humans [29,30]. Consistent with the role of apoE in lipoprotein clearance, transgenic mice expressing human apoE2 - an isoform of apoE defective in receptor binding - are characterized by the presence of β-VLDL (chylomicron and VLDL remnants) and spontaneous atherosclerotic lesions rich in foam cells and displaying small fibrous caps [31].
Besides the obvious role of apoE on cholesterol distribution among lipoproteins, there are several other effects of apoE that might contribute to its anti-atherogenic properties. It has been demonstrated that apoE expression in macrophages is crucial for atherosclerosis protection. Bone marrow transplantation experiments demonstrated that mice receiving macrophages lacking apoE developed significantly more atherosclerosis without affecting total plasma cholesterol [32,33]. ApoE has also been demonstrated to prevent expression of adhesion molecules in human endothelial cells [34] and to inhibit platelet aggregation through the L-argininenitric oxide pathway [35]. Effects on modulation of vascular smooth muscle cell migration and proliferation have been reported as well [36]. Finally, apoE has also been shown to exert a wide variety of immunoregulatory effects: regulation of T cell proliferation and activation [37,38], innate immunity [39], sepsis [40] and more recently macrophage polarization [41].
LDLR KO mouse model
LDLR is a cell surface receptor that recognizes both apoB on LDL and apoE on different lipoproteins. Once lipoproteins bind the LDLR, the complex is internalized removing cholesterol from circulation [42]. Deficiency of LDLR function in humans is basis of familial hypercholesterolemia [43]. Individuals lacking both copies of the LDLR gene present high levels of LDL cholesterol in circulation and suffer myocardial infarction as early as the first decade of life. In 1993, Ishibashi and colleagues reported the generation of LDLR deficient mice [44]. Contrary to the phenotype observed in apoE KO mice, LDLR KO mice do not develop significant atherosclerosis while fed a normal chow diet. This might be explained by the ability of the LDLR-Related Protein (LRP) pathway to clear apoB 48 containing lipoproteins from circulation, such that deletion of LDLR leads only to mild hypercholesterolemia in mice [44,45]. Nevertheless LDLR KO mice are very sensitive to diet modifications. When fed highfat and/or high-cholesterol diets, LDLR KO mice develop severe hypercholesterolemia and atherosclerotic lesions along much of the aortic tree [20,46]. As mentioned previously, humans mostly carry cholesterol on apoB100-containing LDL. To generate animal models with plasma lipoproteins more closely resembling those in humans, Davidson and colleagues generated LDLR KO mice also lacking the apoB mRNA editing catalytic polypeptide-1 (APOBEC-1) gene, rendering them able to synthesize only apoB100 [47], while Hobbs and colleagues generated LDLR KO mice over expressing a human apoB100 transgene [48]. Each of these mice secretes predominantly apoB100, exhibit elevated levels of circulating cholesterol and develop spontaneous atherosclerosis when fed a normal chow diet [47,48].
Advantages of ApoE KO and LDLR KO mouse models of atherosclerosis
The analysis of these two mouse models since their development in the early 1990’s has had a profound impact on our understanding of the molecular pathways involved in atherosclerosis development. Advantages of the apoE and the LDLR KO mouse models of atherosclerosis include the development of complex atherosclerotic plaques in a number of arteries, such as the aortic sinus, aorta, brachiocephalic and carotid arteries [28,49-51]. These complex plaques resemble many features of human atherosclerotic plaques. For example, complex plaques developing in the brachiocephalic artery exhibit some features of plaque rupture; although atherothrombosis is rarely detected [50]. On the other hand, atherosclerosis develops only rarely in coronary arteries of apoE KO or LDLR KO mice, and when it does, generally under extreme conditions. For example, Ishibashi and colleagues detected atherosclerosis in the coronary ostia in LDLR KO mice fed Paigen diet for 7 months [49]. Similarly, atherosclerotic plaques have been detected in coronary arteries of 40 week old apoE KO mice fed a diet containing 21 % fat and 0.15 % cholesterol for 35 weeks [28]. However, myocardial infarction secondary to atherosclerosis, a common feature of human atherosclerotic coronary artery disease, is rarely seen in these mice.
Mouse models of coronary artery disease
ApoE/LDLR double KO (dKO) mice: Mice lacking both apoE and the LDLR develop significant hypercholesterolemia and atherosclerosis in both the aorta and in CA’s in response to long term high fat diet feeding [52]. These mice also developed myocardial infarction in response to hypoxia or mental stress, reportedly mediated by the release of endothelin and consequent vasospasm of partially occluded CA’s, since it was reduced by treatment with an endothelin receptor A blocker [52]. In a separate study, Li and colleagues showed that high-fat diet fed apoE/LDLR dKO mice preconditioned by exposure to either ischemia/reperfusion or hyperoxia presented improved post-ischemic ventricular function and reduced myocardial infarct size [53].
SR-B1/apoE dKO mice: The HDL receptor, scavenger receptor class B type 1 (SR-B1), is a cell surface receptor expressed in several cell types, including hepatocytes, macrophages and endothelial cells, critically involved in atherosclerosis pathogenesis [54]. Transient over expression of SR-B1 in the liver of wild type mice by adenovirus delivery significantly reduced HDL plasma concentrations and increased biliary cholesterol [55]. On the other hand, complete ablation of SR-B1 expression in wild type mice resulted in enlarged HDL particles rich in cholesterol and a decrease in biliary cholesterol [56]. These findings confirmed the importance of this receptor in modulating HDL cholesterol levels in vivo. The process where HDL delivers cholesterol to the liver through SR-B1 is known as reverse cholesterol transport and it is considered a major mechanism through which HDL protects against atherosclerosis development [57]. SR-B1 is a multifunctional protein. It mediates not only the selective transfer of cholesterol esters from HDL into cells, but also the removal of un-esterified cholesterol from cells [58,59]. Moreover, it is also required for HDL signaling in a variety of cells [60,61], best characterized by activation of endothelial Nitric Oxide Synthase (eNOS) in endothelial cells [62]. This process requires apoA1 binding [62]. Activation of eNOS by HDL involved Src-mediated activation of both Akt and mitogen activated protein kinase signaling pathways in an SR-B1 dependent manner [63]. Expression of SR-B1 is required for HDL-mediated inhibition of TNF-a dependent upregulation of expression of the adhesion molecules, Vascular Cell Adhesion Molecule (VCAM)-1 and Intercellular Adhesion Molecule (ICAM)- 1, in endothelial cells, in vitro [54]. VCAM-1 and ICAM-1 play an important role in the accumulation of monocytes in atherosclerotic artery wall. The absence of SR-B1 also affects the anti-oxidant properties of HDL. SR-B1 KO mice were reported to have increased levels of oxidative stress due to a reduction in the circulating levels of HDL-associated antioxidant enzyme paraoxonase-1 [64]. Furthermore, SR-B1 deficiency alters erythrocyte maturation and platelet structure and clearance resulting in the development of anemia and thrombocytopenia [65,66].
The impact of SR-B1 on atherosclerosis development has also been studied in both KO and transgenic models. Hepatic over expression of SR-B1 in heterozygous LDLR KO mice fed a high fat/cholesterol diet, containing the bile salt sodium cholate, decreased lesion area in the aortic root and cholesterol content in VLDL, LDL and HDL [67]. However, protection was not seen in transgenic homozygous LDLR KO fed a high fat/cholesterol diet [67]. On the other hand, transient hepatic over expression of SR-B1 in LDLR KO mice with early and advanced lesions reduced atherosclerosis [68]. Reduction in lesion size was correlated with HDL cholesterol levels [68]. Over expression of SR-B1 in a human apoB transgenic mouse was shown to be protective against diet-induced fatty streak formation in the aorta as well [69]. Krieger and colleagues first reported the effects of complete inactivation of SR-B1 on atherosclerosis [70]. SR-B1 KO mice were crossed with apoE KO mice to generate SR-B1/apoE dKO mice, which were reported to exhibit substantially increased cholesterol associated with VLDL sized particles and abnormally large HDL like particles [70]. When fed normal chow diets, these mice developed accelerated aortic sinus atherosclerosis at 5 weeks of age, at which no atherosclerosis was detected in littermate apoE KO control mice. In a subsequent publication Krieger and colleagues reported that SR-B1/apoE dKO mice fed a normal chow diet developed substantial, occlusive atherosclerosis in CA’s as well, and that this was associated with extensive myocardial fibrosis, cardiomegaly, cardiac conductance abnormalities, reduced cardiac function, and mortality of the dKO mice between 5 and 8 weeks of age (50% mortality by 6 weeks of age) [71].
The nature of the phenotype observed in SR-B1/apoE dKO mice has been further explored. Lymphocyte-dependent inflammation is known to influence atherosclerosis development, thus Karackattu et al. explored a role for T and B cells on CAD development [72]. To do so, RAG-2/SR-B1/apoE triple KO (tKO) mice were generated, where the absence of RAG-2 gene expression prevents lymphocyte maturation [73]. No differences in CA atherosclerosis, myocardial infarction or cardiac dysfunction were found between triple and control SR-B1/ apoE dKO mice, indicating that the absence of circulating B and T lymphocytes does not affect the development of CA atherosclerosis or myocardial infarction, or improve cardiac function in these mice [72]. Hepatic lipase (HL) hydrolyzes triglycerides and phospholipids and it has been suggested that it can influence atherosclerosis, although the mechanism is not clear [74]. Deletion of HL in SR-B1/apoE dKO mice significantly delayed the development of atherosclerosis in both the aortic root and CA’s despite the increase in total cholesterol [75]. This study also confirmed that in these mice the extent of atherosclerosis in CA’s rather than in the aortic root is more closely correlated with cardiac dysfunction and lifespan [75]. Bone marrow transplantation of either wild type or SR-B1/apoE dKO bone marrow into SR-B1/apoE dKO mice significantly reduced serum cholesterol. Transplantation of wild type bone marrow also reduced aortic atherosclerosis and prolonged survival on SR-B1/apoE dKO indicating that restoration of macrophage SR-B1 and/or apoE can reduce atherosclerosis in these dKO mice [76]. In a separate study, microarray mRNA profiling of hearts from SR-B1/apoE dKO mice at different stages of cardiac disease progression revealed a significant increase (80-fold) of apoD in hearts with extensive myocardial infarction [77]. Using a model of ischemia/reperfusion-induced myocardial infarction, Krieger and colleagues showed that apoD could reduce myocardial infarction potentially through its antioxidant activity [77]. This, therefore, led to the identification of apoD as a cardioprotective factor that is induced in hearts upon myocardial infarction.
Pharmacological studies have also been performed in the SRB1/ apoE dKO mice to test the effect of drugs of interest on CAD. Probucol is a potent antioxidant and anti-inflammatory drug reported to prevent atherosclerosis development [78]. Treatment with probucol significantly extended the lifespan of SR-B1/apoE dKO mice (from a mean of 6 to a mean of 36 weeks) and almost completely reversed the cardiac pathology [79]. From this study it was also possible to conclude that most of the pathological features leading towards premature death arise between 3-5 weeks of age, providing a window where the drug could be administered to alter disease progression [79]. In a similar study, SR-B1/apoE dKO mice were treated with either ezetimibe (inhibition of intestinal cholesterol absorption) or SC-435 (inhibition of intestinal bile acid absorption) for three weeks [80]. Both drugs were capable of significantly reducing cholesterol in the IDL/LDL fraction and extending survival of the dKO mice. Treatment with ezetimibe significantly delayed onset or progression of atherosclerosis in the aortic root and CA; myocardial fibrosis and cardiomegaly were reduced as well [80]. Finally, the effect of pomegranate extract, rich in polyphenolic antioxidants, on CAD development was tested [81]. Treatment of SR-B1/apoE dKO mice with pomegranate extract reduced levels of oxidative stress and inflammation in the plaque. CAD and myocardial fibrosis were also reduced, even though treatment increased cholesterol in the VLDL fraction [81]. Despite the reduction in atherosclerosis in the aortic root and CA, survival was not extended in SR-B1/apoE dKO mice receiving pomegranate extract.
SR-B1KO/apoE-hypomorphic mice: ApoE-hypomorphic mice are characterized by a reduced expression- around 5% of normal- of a mutant form of apoE (Arg-61 allelic variant, resembling human apoE4) [82]. The hypomorphic expression of the mutant form of apoE was caused by the insertion of a neomycin cassette flanked by loxP sites into the third intron of the apoE gene [82]. Reduced expression of apoE, however, does not affect lipoprotein cholesterol profiles, with most cholesterol present in the HDL fraction, when mice were fed a normal chow diet [83]. When challenged with a high fat diet, apoE-hypomorphic mice rapidly develop hypercholesterolemia characterized by increased cholesterol associated with VLDL and IDL/LDL sized lipoproteins and reduced cholesterol associated with HDL. The hypercholesterolemia could be fully reverted by normal chow diet feeding [83]. Crossing apoE-hypomorphic mice with SR-B1 KO mice generated the SR-B1KO/apoE-hypomorphic mouse model [84]. When fed a normal chow diet, SR-B1KO/apoE-hypomorphic mice are healthy with no signs of CAD or hypercholesterolemia [84]. However, when SR-B1KO/apoE-hypomorphic mice are challenged with a Paigen diet, they develop severe hypercholesterolemia, aortic root atherosclerosis and CAD, displaying a phenotype similar to that of SR-B1/apoE dKO mice [84]. Further studies by Krieger and colleagues also revealed that the rate of disease progression could be altered by changing the severity of the atherogenic diet and accelerated by social isolation [85]. Hence, the SR-B1KO/apoE-hypomorphic mouse model has the advantage of diet-induced atherosclerosis allowing for the study of other conditions that may affect disease development besides diet.
Bone marrow transplantation experiments allow for evaluation of the impact of different macrophage genes on plaque development. Using this approach, Pei and colleagues set out to explore the role of macrophage SR-B1 in atherosclerosis, taking advantage of the inducible nature of plaque formation in SR-B1KO/apoE-hypomorphic mice [86]. Transplantation of SR-B1KO/apoE-hypomorphic mice with SR-B1+/+ bone marrow restored SR-B1 expression in macrophages and reduced the development of diet-induced atherosclerosis in both the aortic sinus and coronary arteries with a consequent reduction in cardiac fibrosis. The reduction was attributed mainly to a reduction in monocyte recruitment into plaques in mice with SR-B1 expression restored in bone marrow derived cells [86].
The SR-B1KO/apoE-hypomorphic mouse model has also been used to explore the potential of drugs to treat CVD. Raffai and colleagues evaluated the effect of FTY720 administration on ischemia/reperfusion injury in hearts ex vivo and on diet-induced coronary heart disease in SR-B1KO/apoE-hypomorphic mice [87]. FTY720 is an analogue of sphingosine-1-phosphate, able to enhance cardiomyocyte survival in hypoxia, to prevent arrhythmias as a consequence of ischemia/reperfusion injury and to protect against atherosclerosis development in both apoE and LDLR KO mice [88]. Moreover, FTY720 is also well-known immune modulator, causing lymphopenia due to impaired lymphocyte trafficking from lymph nodes into circulation [88]. Long-term treatment of wild type mice with FTY720 significantly protected the heart from damage induced by ex vivo ischemia/reperfusion, and treatment of high fat/high cholesterol diet-fed SR-B1KO/apoE-hypomorphic mice with FTY720 improved their survival and protected against cardiac dysfunction [87]. The cardioprotection seen was not accompanied by protection against atherosclerosis development; rather it was attributed to immunosuppression leading to reduced cardiac remodeling [87]. It is noteworthy that the effect of FTY720 treatment of SR-B1KO/apoEhypomorphic mice differs from the phenotype seen in RAG2/SRB1/ apoE tKO where no differences were detected in atherosclerosis and cardiac function [72]. Although the models are different, this discordance suggests that FTY720 might exert protective effects through other mechanisms in addition to induction of lymphopenia. In a more recent publication [89], Raffai and colleagues made use of the diet inducible nature of atherosclerosis and myocardial infarction in the SR-B1KO/apoE-hypomorphic mice to examine the effects of FTY720 specifically on cardiac failure subsequent to CA atherosclerosis and myocardial infarction. To do this, they first fed SR-B1KO/apoE-hypomorphic mice a high fat, high cholesterol diet for 3.5 weeks to induce CA atherosclerosis and myocardial infarction. For those mice that survived the feeding, they then switched them to a normal chow diet and corrected the apoE-hypomorphic mutation by Cre-mediated excision of the inserted neomycin gene cassette from the apoE locus, to reduce hypercholesterolemia, and began treatment with FTY720. Reduction of hypercholesterolemia alone, post infarction, did not improve cardiac function. On the other hand, FTY720 treatment post infarction was able to reverse cardiac dysfunction. This was associated with a reduction in the number of B cells and the expression of matrix metalloproteinase-2 and inflammatory genes in the heart. This study demonstrates that this and other diet inducible models of coronary artery atherosclerosis and myocardial infarction (see below) may be useful model systems to study the post-infarction development of heart failure.
SR-B1/LDLR dKO mice: SR-B1/LDLR dKO mice challenged with a high fat, but relatively low (0.15%) cholesterol atherogenic diet for two months exhibit increased atherosclerosis in the aortic root when compared with LDLR KO mice [90]. They also present most cholesterol associated with large HDL particles and reduced apoB levels in the IDL/LDL fraction but no CA atherosclerosis. Previous studies in LDLR KO mice with attenuated expression of SR-B1 also showed an increase in atherosclerosis development when mice were challenged with a high fat diet, however in these mice increased cholesterol was mostly associated with LDL-sized particles [91]. The cholesterol distribution seen in dKO mice likely suggests that impaired HDL cholesterol clearance contributes to atherosclerosis development [90].
Further characterization of the SR-B1/LDLR dKO mice revealed that the occurrence of CA atherosclerosis is dependent on the cholesterol and cholate content of the diets [92]. SR-B1/LDLR dKO mice fed diets rich in cholesterol with or without cholate developed high levels of occlusive CA atherosclerosis along with myocardial infarctions and reduced survival [92]. SR-B1/LDLR dKO mice fed a Western type (high fat, relatively low, 0.15 % cholesterol) diet developed less occlusive CA disease, and this did not result in significant myocardial fibrosis or reduction of survival. High fat/ cholesterol diet-dependent platelet accumulation in the CA was also observed, with the highest levels associated with the presence of cholate in the diet [92], suggesting the contribution of thrombosis on top of atherosclerosis to occlusive coronary artery disease. The SR-B1/LDLR dKO mice also exhibited higher levels of circulating cytokines, monocytosis than control LDLR single KO mice. Finally, CA’s from atherogenic diet fed SR-B1/LDLR dKO mice also expressed substantially higher levels of ICAM-1 and VCAM-1 than similarly fed control LDLR single KO mice, which might explain the increased susceptibility of CA’s from SR-B1 deficient mice to atherosclerosis development, compared to CA’s from SR-B1 expressing mice [92].diet developed less occlusive CA disease, and this did not result in significant myocardial fibrosis or reduction of survival. High fat/ cholesterol diet-dependent platelet accumulation in the CA was also observed, with the highest levels associated with the presence of cholate in the diet [92], suggesting the contribution of thrombosis on top of atherosclerosis to occlusive coronary artery disease. The SR-B1/LDLR dKO mice also exhibited higher levels of circulating cytokines, monocytosis than control LDLR single KO mice. Finally, CA’s from atherogenic diet fed SR-B1/LDLR dKO mice also expressed substantially higher levels of ICAM-1 and VCAM-1 than similarly fed control LDLR single KO mice, which might explain the increased susceptibility of CA’s from SR-B1 deficient mice to atherosclerosis development, compared to CA’s from SR-B1 expressing mice [92].
Advantages of SR-B1 Deficient Mouse Models of Coronary Artery Atherosclerosis
Together, these SR-B1 deficient mice (on the various atherogenic backgrounds detailed above) have a number of advantages. Like the apoE KO and LDLR KO mice, they appear to develop large and complex atherosclerotic plaques, at least in the aortic sinus [92]. Furthermore, the rapid development of atherosclerosis in CA’s, characterized by the accumulation of platelets and accompanied by myocardial fibrosis suggests that these compound mutant mice recapitulate many features of human atherosclerotic coronary heart disease. The rapid development of coronary artery atherosclerosis and myocardial fibrosis, which is spontaneous in the SR-B1/apoE dKO mice, and rapidly induced (3.5 weeks) by high fat, high cholesterol diets containing cholate in the SR-B1 KO/apoE-hypomorphic and SR-B1/LDLR dKO mice, may be an asset for rapid pre-clinical testing of novel therapeutics.
Other Gene Targeted Mice that Develop CA Atherosclerosis
PDZK1/apoE dKO mice: PDZK1 (PDZ containing 1) was identified as a cytoplasmic adaptor that interacts with a number of membrane-associated proteins, including cell surface receptors and ion channels, via its four PDZ (Postsynaptic density protein-95, Drosophila disc-large protein, Zonula occludens protein1) proteinprotein interaction domains [93-96]. In 2000, Ikemoto and colleagues first showed that PDZK1 binds to the C-terminus of SRB1, via its first PDZ domain, in the sinusoidal membranes of rat liver [97]. Kocher, Krieger and colleagues further characterized this interaction both in vitro and in vivo and explored the lower affinity binding of the C terminus of SR-B1 to the third PDZ domain [98, 99]. The inactivation of PDZK1 expression in mice caused a 95% decrease in SR-B1 protein abundance in liver and 50% decrease in intestine; in contrast, SR-B1 protein levels were unaffected in other tissues including adrenal tissue, endothelial cells and macrophages [100-103]. Hepatic overexpression of transgenes encoding all four domains of PDZK1 could restore SR-B1 expression and function in the liver [104, 105]. As expected, PDZK1 KO mice showed increased plasma total cholesterol, HDL-C and HDL particle size, similar to the phenotypes observed in SR-B1 KO mice [101]. Global knockout of PDZK1 in apoE KO mice (PDZK1/apoE dKO) resulted in increased high fat diet-induced aortic atherosclerosis, but no occlusive CA atherosclerosis, myocardial infarction or early death [102]. However when these mice were fed a more atherogenic, Paigen diet, containing higher cholesterol as well as sodium cholate, they developed 26% increased atherosclerosis in aortic sinus as well as lipid-rich plaques in CA’s among which about 40% were completely occluded [106]. In addition, trichrome staining showed that these PDZK1/apoE dKO mice fed the Paigen diet developed extensive cardiac fibrosis [106], resembling the CA atherosclerosis and myocardial fibrosis of SR-B1/ apoE dKO mice (which, as indicated above, occurred spontaneously). On the other hand, unlike the case for SR-B1/apoE dKO mice, the Paigen diet fed PDZK1/apoE dKO mice did not develop cardiomegaly or reduced lifespan over the feeding time course analyzed [106]. Thus, PDZK1/apoE dKO mice exhibit less severe atherosclerotic disease compared to SR-B1/apoE dKO mice, possibly reflecting partial protection due to SR-B1 expressed outside of the liver. On the other hand the resemblance of the coronary artery atherosclerosis phenotypes in SR-B1/apoE dKO and PDZK/apoE dKO mice may reflect a role for PDZK1 in SR-B1’s function outside of the liver. Even though deficiency of PDZK1 did not affect SR-B1 expression in vascular cells, such as macrophages and endothelial cells which play vital roles in the development of atherosclerosis, PDZK1 has been shown to play an important role in SR-B1 dependent signaling in response to HDL in these cells [100,103,107]. On the other hand, it has recently been shown that inactivation of PDZK1 results in increased vascular smooth muscle cell proliferation and migration in vitro and neointima formation in vivo in mice, via an apparently SR-B1 independent pathway [108]. Immunohistochemical staining revealed little evidence of smooth muscle contribution to the lipid rich aortic sinus atherosclerotic plaques in Paigen diet fed PDZK1/ apoE dKO mice, however the contribution of smooth muscle cells to occlusive coronary artery atherosclerotic plaques was not described [106]. The coronary artery atherosclerotic phenotypes of PDZK1 KO mice are likely to be influenced by both the altered lipid profile due to the knockdown of hepatic SR-B1, as well as the impaired SR-B1- mediated signaling pathway in vascular cells. The contribution of PDZK1 in smooth muscle cells to protection against coronary artery atherosclerosis is currently unclear.
eNOS/apoE dKO mice: Nitric oxide is a potent vasodilator whose availability is dramatically decreased in hypercholesterolemiainduced vascular disease [109]. ApoE KO mice deficient in eNOS develop increased atherosclerosis compared to controls when fed a Western diet for 4-6 months [110]. eNOS/apoE dKO mice also developed CA atherosclerosis, aortic aneurysm and showed evidence of myocardial ischemia and left ventricle dysfunction [110]. No effects on survival were reported. eNOS/apoE dKO mice also showed a significant increase in blood pressure, however treatment of mice with hydralazine (a blood pressure lowering drug) did not affect atherosclerosis or aortic aneurysm formation, indicating that hypertension is not required for atherosclerosis development in these animals [111]. SR-B1 has been shown to mediate HDL signaling in endothelial cells, leading to activation of eNOS reviewed in [107]. As for PDZK1/apoE KO mice, the similarities in the CA atherosclerosis phenotypes between the SR-B1/apoE dKO and the eNOS/apoE dKO mice may suggest a common pathway involved in protection against CA atherosclerosis.
AKT1/apoE dKO mice: The AKT family of kinases is involved in several cell functions including regulation of proliferation, metabolism and cell survival in several cells types including those in the cardiovascular system [112]. Genetic deletion of AKT isoform 1 (AKT1) led to increased atherosclerosis in apoE KO mice fed a high cholesterol diet for 14 weeks [113]. Lesions were found in the aortic sinus, aorta and in CA’s. This was accompanied by increased apoptosis in both endothelial cells and macrophages and decrease phosphorylation of eNOS. The absence of AKT1 was also characterized by increased macrophage abundance and proinflammatory markers in the vessel wall, which may contribute to lesion formation [113]. More in depth analysis of plaque composition showed presence of increased necrotic core sizes and reduction in fibrous cap and collagen content in dKO animals [114]. Loss of AKT1 also led to myocardial infarction, cardiac dysfunction and significant increase in mortality [113,114]. Surprisingly, the inactivation of AKT1 on an SR-B1/apoE dKO background led to alleviation of several of the pathological features of these mice including aortic root and coronary artery atherosclerosis, cardiac dysfunction, myocardial fibrosis and improved survival [115]. Protection in AKT1/SR-B1/apoE tKO mice seems to be associated to an overall reduction in oxidative stress, with reductions in lipid oxidation and foam cell formation [115]. These results are surprising given that inactivation of either SR-B1 or AKT1 individually on an apoE KO background lead to spontaneous or diet induced (respectively) CA atherosclerosis and myocardial infarction and reduced lifespan. Further studies are needed to clarify this apparently dual role of AKT1 on atherosclerotic coronary heart disease development.
uPA-Tg/apoE KO mice: Urokinase type Plasminogen Activator (uPA) is a serine protease involved in plasmin generation [116]. uPA is expressed by macrophages in the wall of human arteries and the level of expression correlates with the severity of the disease in CA’s [116]. Transgenic mice overexpressing uPA in macrophages (uPA-Tg), when crossed to ApoE KO mice developed larger high fat diet induced plaques in the aorta when fed for 10 weeks but exhibited no differences in plaque structure [117]. ApoE KO mice overexpressing uPA in macrophages also developed high fat diet induced CA atherosclerosis and myocardial infarction and exhibited early mortality. The phenotype seen in the uPA-Tg/ApoE KO mice is dependent on the presence of plasminogen (Plg) since macrophage overexpression of uPA had no effect on atherosclerosis development in Plg/ApoE dKO mice [118]. Interestingly, overexpression of macrophage uPA in otherwise wild type mice increased macrophage accumulation and cardiac fibrosis in the absence of atherosclerosis [119]. The mechanism behind cardiac fibrosis seems to be dependent on plasminogen and plasmin activation, but the identity of the plasmin substrate(s) that may contribute to the phenotype remains unclear [120].
Conclusion
In this review, we aimed to summarize the main features of the current mouse models of CAD with particular emphasis on those involving inactivation of SR-B1. The use of the mouse to understand the pathology and progression of atherosclerosis has been expanded as a result of the advances in genetic engineering techniques. Easy manipulation and the low cost of maintenance have placed the mouse on top of other animal models like rabbits and non-human primates. Since the development of the ApoE and LDLR KO mouse models, the impact of several genes and signaling pathways on atherosclerosis has been explored increasing our understanding of disease pathophysiology. The introduction of mouse models that can now emulate features of human atherosclerosis -including CA atherosclerosis and myocardial infarction, with special emphasis in those deficient on SR-B1, represent an opportunity to further explore disease mechanisms. The phenotype of compound mutant mice lacking SR-B1 together with other atherosclerosis predisposing mutations strongly resembles certain characteristics of human CAD and seems to be multifactorial involving multiple cell types. Further clarification on the role of SR-B1 on these cell types can potentially generate new targets for the development of treatments to prevent or treat atherosclerosis.
Acknowledgement
LG received a graduate scholarship from the Equal Opportunities Canada-Chile Program. PY received a graduate scholarship from the China Scholarship Council. This work was supported by research funding to BLT from the Canadian Institutes for Health Research (MOP74765) and the Heart and Stroke Foundation of Canada (G- 15-0009016).
References
- Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001; 104: 503-516.
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011; 473: 317-325.
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation. 2015; 131: e29-322.
- Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011; 123: 933-944.
- Yach D, Hawkes C, Gould CL, Hofman KJ. The global burden of chronic diseases: overcoming impediments to prevention and control. JAMA. 2004; 291: 2616-2622.
- Tanganelli P, Bianciardi G, Simoes C, Attino V, Tarabochia B, Weber G. Distribution of lipid and raised lesions in aortas of young people of different geographic origins (WHO-ISFC PBDAY Study). World Health Organization-International Society and Federation of Cardiology. Pathobiological Determinants of Atherosclerosis in Youth. Arteriosclerosis and thrombosis: a journal of vascular biology / American Heart Association. 1993; 13: 1700-1710.
- Kiechl S, Willeit J. The natural course of atherosclerosis. Part I: incidence and progression. Arterioscler Thromb Vasc Biol. 1999; 19: 1484-1490.
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006; 6: 508-519.
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007; 7: 678-689.
- McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011; 50: 331-347.
- Newby AC, Zaltsman AB. Fibrous cap formation or destruction--the critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc Res. 1999; 41: 345-360.
- Halvorsen B, Otterdal K, Dahl TB, Skjelland M, Gullestad L, Øie E, et al. Atherosclerotic plaque stability--what determines the fate of a plaque? Prog Cardiovasc Dis. 2008; 51: 183-194.
- Tanzawa K, Shimada Y, Kuroda M, Tsujita Y, Arai M, Watanabe H. WHHL-rabbit: a low density lipoprotein receptor-deficient animal model for familial hypercholesterolemia. FEBS Lett. 1980; 118: 81-84.
- Griggs TR, Bauman RW, Reddick RL, Read MS, Koch GG, Lamb MA. Development of coronary atherosclerosis in swine with severe hypercholesterolemia. Lack of influence of von Willebrand factor or acute intimal injury. Arteriosclerosis. 1986; 6: 155-165.
- Faggiotto A, Ross R, Harker L. Studies of hypercholesterolemia in the nonhuman primate. I. Changes that lead to fatty streak formation. Arteriosclerosis. 1984; 4: 323-340.
- de Silva HV, Más-Oliva J, Taylor JM, Mahley RW. Identification of apolipoprotein B-100 low density lipoproteins, apolipoprotein B-48 remnants, and apolipoprotein E-rich high density lipoproteins in the mouse. J Lipid Res. 1994; 35: 1297-1310.
- Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 1977; 62: 707-714.
- Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985; 57: 65-73.
- Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and HDL-lipid levels in male, female, and testosterone-treated female mice from strains C57BL/6, BALB/c, and C3H. Atherosclerosis. 1987; 64: 215-221.
- Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995; 36: 2320-2328.
- Mehrabian M, Demer LL, Lusis AJ. Differential accumulation of intimal monocyte-macrophages relative to lipoproteins and lipofuscin corresponds to hemodynamic forces on cardiac valves in mice. Arteriosclerosis and thrombosis: a journal of vascular biology / American Heart Association. 1991; 11: 947-957.
- Liao F, Andalibi A, deBeer FC, Fogelman AM, Lusis AJ. Genetic control of inflammatory gene induction and NF-kappa B-like transcription factor activation in response to an atherogenic diet in mice. J Clin Invest. 1993; 91: 2572-2579.
- Imaizumi K. Diet and atherosclerosis in apolipoprotein E-deficient mice. Biosci Biotechnol Biochem. 2011; 75: 1023-1035.
- Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992; 71: 343-353.
- Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992; 89: 4471-4475.
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992; 258: 468-471.
- Ghiselli G, Schaefer EJ, Gascon P, Breser HB Jr. Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science. 1981; 214: 1239-1241.
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994; 14: 133-140.
- Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994; 14: 141-147.
- Zhang SH, Reddick RL, Burkey B, Maeda N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994; 94: 937-945.
- Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest. 1998; 102: 130-135.
- Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, et al. Increased atherosclerosis in mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci USA. 1997; 94: 4647-4652.
- Van Eck M, Herijgers N, Yates J, Pearce NJ, Hoogerbrugge PM, Groot PH, et al. Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, (beta)VLDL catabolism, and atherosclerosis. Arterioscler Thromb Vasc Biol. 1997; 17: 3117-3126.
- Stannard AK, Riddell DR, Sacre SM, Tagalakis AD, Langer C, von Eckardstein A, et al. Cell-derived apolipoprotein E (ApoE) particles inhibit vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J Biol Chem. 2001; 276: 46011-46016.
- Riddell DR, Graham A, Owen JS. Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway. Implications for vascular disease. J Biol Chem. 1997; 272: 89-95.
- Swertfeger DK, Hui DY. Apolipoprotein E receptor binding versus heparan sulfate proteoglycan binding in its regulation of smooth muscle cell migration and proliferation. J Biol Chem. 2001; 276: 25043-25048.
- Zhang HL, Wu J, Zhu J. The immune-modulatory role of apolipoprotein E with emphasis on multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Dev Immunol. 2010; 186813.
- Kelly ME, Clay MA, Mistry MJ, Hsieh-Li HM, Harmony JA. Apolipoprotein E inhibition of proliferation of mitogen-activated T lymphocytes: production of interleukin 2 with reduced biological activity. Cell Immunol. 1994; 159: 124-139.
- Roselaar SE, Daugherty A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res. 1998; 39: 1740-1743.
- Kattan OM, Kasravi FB, Elford EL, Schell MT, Harris HW. Apolipoprotein E-mediated immune regulation in sepsis. J Immunol. 2008; 181: 1399-1408.
- Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011; 31: 1160-1168.
- Goldstein JL, Brown MS. Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987; 76: 504-507.
- Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979; 13: 259-289.
- Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993; 92: 883-893.
- Wouters K, Shiri-Sverdlov R, van Gorp PJ, van Bilsen M, Hofker MH. Understanding hyperlipidemia and atherosclerosis: lessons from genetically modified apoe and ldlr mice. Clin Chem Lab Med. 2005; 43: 470-479.
- Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol. 1999; 19: 1938-1944.
- Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, et al. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998; 4: 934-938.
- Sanan DA, Newland DL, Tao R, Marcovina S, Wang J, Mooser V, et al. Low density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein(a). Proc Natl Acad Sci USA. 1998; 95: 4544-4549.
- Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994; 93: 1885-1893.
- Matoba T, Sato K, Egashira K. Mouse models of plaque rupture. Curr Opin Lipidol. 2013; 24: 419-425.
- VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004; 24: 12-22.
- Caligiuri G, Levy B, Pernow J, Thorén P, Hansson GK. Myocardial infarction mediated by endothelin receptor signaling in hypercholesterolemic mice. Proc Natl Acad Sci USA. 1999; 96: 6920-6924.
- Li G, Tokuno S, Tähep ld P, Vaage J, Löwbeer C, Valen G. Preconditioning protects the severely atherosclerotic mouse heart. Ann Thorac Surg. 2001; 71: 1296-1303.
- Al-Jarallah A, Trigatti BL. A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochim Biophys Acta. 2010; 1801: 1239-1248.
- Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 1997; 387: 414-417.
- Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci USA. 1997; 94: 12610-12615.
- Trigatti BL, Krieger M, Rigotti A. Influence of the HDL receptor SR-BI on lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003; 23: 1732-1738.
- Brundert M, Ewert A, Heeren J, Behrendt B, Ramakrishnan R, Greten H, et al. Scavenger receptor class B type I mediates the selective uptake of high-density lipoprotein-associated cholesteryl ester by the liver in mice. Arterioscler Thromb Vasc Biol. 2005; 25: 143-148.
- Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, et al. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998; 273: 5599-5606.
- Mineo C, Shaul PW. Regulation of signal transduction by HDL. J Lipid Res. 2013; 54: 2315-2324.
- Nofer JR. Signal transduction by HDL: agonists, receptors, and signaling cascades. Handb Exp Pharmacol. 2015; 224: 229-256.
- Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001; 7: 853-857.
- Mineo C, Yuhanna IS, Quon MJ, Shaul PW. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003; 278: 9142-9149.
- Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK, et al. Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol. 2007; 27: 2413-2419.
- Holm TM, Braun A, Trigatti BL, Brugnara C, Sakamoto M, Krieger M, et al. Failure of red blood cell maturation in mice with defects in the high-density lipoprotein receptor SR-BI. Blood. 2002; 99: 1817-1824.
- Dole VS, Matuskova J, Vasile E, Yesilaltay A, Bergmeier W, Bernimoulin M, et al. Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008; 28: 1111-1116.
- Arai T, Wang N, Bezouevski M, Welch C, Tall AR. Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J Biol Chem. 1999; 274: 2366-2371.
- Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000; 20: 721-727.
- Ueda Y, Gong E, Royer L, Cooper PN, Francone OL, Rubin EM. Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J Biol Chem. 2000; 275: 20368-20373.
- Trigatti B, Rayburn H, Vi&nTilde;als M, Braun A, Miettinen H, Penman M, et al. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA. 1999; 96: 9322-9327.
- Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, et al. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002; 90: 270-276.
- Karackattu SL, Picard MH, Krieger M. Lymphocytes are not required for the rapid onset of coronary heart disease in scavenger receptor class B type I/apolipoprotein E double knockout mice. Arterioscler Thromb Vasc Biol. 2005; 25: 803-808.
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992; 68: 855-867.
- Jansen H, Verhoeven AJ, Sijbrands EJ. Hepatic lipase: a pro- or anti-atherogenic protein? J Lipid Res. 2002; 43: 1352-1362.
- Karackattu SL, Trigatti B, Krieger M. Hepatic lipase deficiency delays atherosclerosis, myocardial infarction, and cardiac dysfunction and extends lifespan in SR-BI/apolipoprotein E double knockout mice. Arterioscler Thromb Vasc Biol. 2006; 26: 548-554.
- Yu H, Zhang W, Yancey PG, Koury MJ, Zhang Y, Fazio S, et al. Macrophage apolipoprotein E reduces atherosclerosis and prevents premature death in apolipoprotein E and scavenger receptor-class BI double-knockout mice. Arterioscler Thromb Vasc Biol. 2006; 26: 150-156.
- Tsukamoto K, Mani DR, Shi J, Zhang S, Haagensen DE, Otsuka F, et al. Identification of apolipoprotein D as a cardioprotective gene using a mouse model of lethal atherosclerotic coronary artery disease. Proc Natl Acad Sci USA. 2013; 110: 17023-17028.
- Yamashita S, Matsuzawa Y. Where are we with probucol: a new life for an old drug? Atherosclerosis. 2009; 207: 16-23.
- Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, et al. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci USA. 2003; 100: 7283-7288.
- Braun A, Yesilaltay A, Acton S, Broschat KO, Krul ES, Napawan N, et al. Inhibition of intestinal absorption of cholesterol by ezetimibe or bile acids by SC-435 alters lipoprotein metabolism and extends the lifespan of SR-BI/apoE double knockout mice. Atherosclerosis. 2008; 198: 77-84.
- Al-Jarallah A, Igdoura F, Zhang Y, Tenedero CB, White EJ, MacDonald ME, et al. The effect of pomegranate extract on coronary artery atherosclerosis in SR-BI/APOE double knockout mice. Atherosclerosis. 2013; 228: 80-89.
- Raffai RL, Dong LM, Farese RV Jr, Weisgraber KH. Introduction of human apolipoprotein E4 "domain interaction" into mouse apolipoprotein E. Proc Natl Acad Sci USA. 2001; 98: 11587-11591.
- Raffai RL, Weisgraber KH. Hypomorphic apolipoprotein E mice: a new model of conditional gene repair to examine apolipoprotein E-mediated metabolism. J Biol Chem. 2002; 277: 11064-11068.
- Zhang S, Picard MH, Vasile E, Zhu Y, Raffai RL, Weisgraber KH, et al. Diet-induced occlusive coronary atherosclerosis, myocardial infarction, cardiac dysfunction, and premature death in scavenger receptor class B type I-deficient, hypomorphic apolipoprotein ER61 mice. Circulation. 2005; 111: 3457-3464.
- Nakagawa-Toyama Y, Zhang S, Krieger M. Dietary manipulation and social isolation alter disease progression in a murine model of coronary heart disease. PloS one. 2012; 7: e47965.
- Pei Y, Chen X, Aboutouk D, Fuller MT, Dadoo O, Yu P, et al. SR-BI in bone marrow derived cells protects mice from diet induced coronary artery atherosclerosis and myocardial infarction. PLoS One. 2013; 8: e72492.
- Wang G, Kim RY, Imhof I, Honbo N, Luk FS, Li K, et al. The immunosuppressant FTY720 prolongs survival in a mouse model of diet-induced coronary atherosclerosis and myocardial infarction. Journal of cardiovascular pharmacology. 2014; 63: 132-143.
- Karliner JS. Sphingosine kinase and sphingosine 1-phosphate in the heart: a decade of progress. Biochim Biophys Acta. 2013; 1831: 203-212.
- Luk FS, Kim RY, Li K, Ching D, Wong DK, Joshi SK, et al. Immunosuppression with FTY720 Reverses Cardiac Dysfunction in Hypomorphic ApoE Mice Deficient in SR-BI Expression that Survive Myocardial Infarction Caused by Coronary Atherosclerosis. Journal of cardiovascular pharmacology. 2016.
- Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2003; 23: 1589-1594.
- Huszar D, Varban ML, Rinninger F, Feeley R, Arai T, Fairchild-Huntress V, et al. Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arterioscler Thromb Vasc Biol. 2000; 20: 1068-1073.
- Fuller M, Dadoo O, Serkis V, Abutouk D, MacDonald M, Dhingani N, et al. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol. 2014; 34: 2394-2403.
- Kocher O, Pal R, Roberts M, Cirovic C, Gilchrist A. Targeted disruption of the PDZK1 gene by homologous recombination. Mol Cell Biol. 2003; 23: 1175-1180.
- Anzai N, Miyazaki H, Noshiro R, Khamdang S, Chairoungdua A, Shin HJ, et al. The multivalent PDZ domain-containing protein PDZK1 regulates transport activity of renal urate-anion exchanger URAT1 via its C terminus. J Biol Chem. 2004; 279: 45942-45950.
- Lenzen H, Lünnemann M, Bleich A, Manns MP, Seidler U, Jörns A. Downregulation of the NHE3-binding PDZ-adaptor protein PDZK1 expression during cytokine-induced inflammation in interleukin-10-deficient mice. PLoS One. 2012; 7: e40657.
- Kocher O, Comella N, Tognazzi K, Brown LF. Identification and partial characterization of PDZK1: a novel protein containing PDZ interaction domains. Lab Invest. 1998; 78: 117-125.
- Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, et al. Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc Natl Acad Sci USA. 2000; 97: 6538-6543.
- Kocher O, Birrane G, Yesilaltay A, Shechter S, Pal R, Daniels K, et al. Identification of the PDZ3 domain of the adaptor protein PDZK1 as a second, physiologically functional binding site for the C terminus of the high density lipoprotein receptor scavenger receptor class B type I. J Biol Chem. 2011; 286: 25171-25186.
- Kocher O, Birrane G, Tsukamoto K, Fenske S, Yesilaltay A, Pal R, et al. In vitro and in vivo analysis of the binding of the C terminus of the HDL receptor scavenger receptor class B, type I (SR-BI), to the PDZ1 domain of its adaptor protein PDZK1. J Biol Chem. 2010; 285: 34999-35010.
- Al-Jarallah A, Chen X, Gonzalez L, Trigatti BL. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PloS one. 2014; 9: e106487.
- Kocher O, Yesilaltay A, Cirovic C, Pal R, Rigotti A, Krieger M. Targeted disruption of the PDZK1 gene in mice causes tissue-specific depletion of the high density lipoprotein receptor scavenger receptor class B type I and altered lipoprotein metabolism. J Biol Chem. 2003; 278: 52820-52825.
- Kocher O, Yesilaltay A, Shen CH, Zhang S, Daniels K, Pal R, et al. Influence of PDZK1 on lipoprotein metabolism and atherosclerosis. Biochim Biophys Acta. 2008; 1782: 310-316.
- Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. 2008; 102: 480-487.
- Fenske SA, Yesilaltay A, Pal R, Daniels K, Barker C, Quinones V, et al. Normal hepatic cell surface localization of the high density lipoprotein receptor, scavenger receptor class B, type I, depends on all four PDZ domains of PDZK1. J Biol Chem. 2009; 284: 5797-5806.
- Fenske SA, Yesilaltay A, Pal R, Daniels K, Rigotti A, Krieger M, et al. Overexpression of the PDZ1 domain of PDZK1 blocks the activity of hepatic scavenger receptor, class B, type I by altering its abundance and cellular localization. J Biol Chem. 2008; 283: 22097-22104.
- Yesilaltay A, Daniels K, Pal R, Krieger M, Kocher O. Loss of PDZK1 causes coronary artery occlusion and myocardial infarction in Paigen diet-fed apolipoprotein E deficient mice. PLoS One. 2009; 4: e8103.
- Saddar S, Mineo C, Shaul PW. Signaling by the high-affinity HDL receptor scavenger receptor B type I. Arterioscler Thromb Vasc Biol. 2010; 30: 144-150.
- Lee WR, Sacharidou A, Behling-Kelly E, Oltmann SC, Zhu W, Ahmed M, et al. PDZK1 prevents neointima formation via suppression of breakpoint cluster region kinase in vascular smooth muscle. PLoS One. 2015; 10: e0124494.
- Shaul PW. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol. 2003; 547: 21-33.
- Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001; 104: 448-454.
- Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation. 2001; 104: 2391-2394.
- Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascul Pharmacol. 2015; 74: 38-48.
- Fernández-Hernando C, Ackah E, Yu J, Suárez Y, Murata T, Iwakiri Y, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007; 6: 446-457.
- Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, Sessa WC. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009; 29: 2033-2040.
- Kerr BA, Ma L, West XZ, Ding L, Malinin NL, Weber ME, et al. Interference with akt signaling protects against myocardial infarction and death by limiting the consequences of oxidative stress. Sci Signal. 2013; 6: ra67.
- Kienast J, Padro T, Steins M, Li CX, Schmid KW, Hammel D, et al. Relation of urokinase-type plasminogen activator expression to presence and severity of atherosclerotic lesions in human coronary arteries. Thromb Haemost. 1998; 79: 579-586.
- Cozen AE, Moriwaki H, Kremen M, DeYoung MB, Dichek HL, Slezicki KI, et al. Macrophage-targeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death. Circulation. 2004; 109: 2129-2135.
- Kremen M, Krishnan R, Emery I, Hu JH, Slezicki KI, Wu A, et al. Plasminogen mediates the atherogenic effects of macrophage-expressed urokinase and accelerates atherosclerosis in apoE-knockout mice. Proc Natl Acad Sci USA. 2008; 105: 17109-17114.
- Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res. 2004; 95: 637-644.
- Stempien-Otero A, Plawman A, Meznarich J, Dyamenahalli T, Otsuka G, Dichek DA. Mechanisms of cardiac fibrosis induced by urokinase plasminogen activator. J Biol Chem. 2006; 281: 15345-15351.