
Research Article
J Cardiovasc Disord. 2021; 7(1): 1041.
Epigallocatechin Gallate (EGCG) – A Novel Covalent NF- κB Inhibitor: Structural and Molecular Characterization
Reddy AT*, Lakshmi SP, Varadacharyulu N.Ch and Kodidhela LD
Department of Biochemistry, Sri Krishnadevaraya University, Anantapur, Andhra Pradesh, India
*Corresponding author: Aravind T Reddy, Department of Biochemistry, Sri Krishnadevaraya University, Anantapur, AP, India
Received: May 08, 2021; Accepted:May 31, 2021; Published: June 07, 2021
Abstract
Tea contains antioxidant catechins thought to exert health-promoting protective effects against conditions involving chronic inflammation, such as cardiovascular diseases. The most abundant catechin in tea is Epigallocatechin Gallate (EGCG), thought to be a key contributor to tea’s health-promoting actions. EGCG exerts protective cardiovascular effects via its antioxidant, antiinflammatory, hypolipidemic, anti-thrombogenic, and anti-hypertensive actions. Because EGCG inhibits the strong proinflammatory gene-inducing transcription factor NF-κB, we analyzed the chemical and molecular details of the mechanism by which EGCG mediates NF-κB inhibition. We quantified and mapped key parameters of its chemical reactivity including its electrophilic Fukui ƒ+ function, in silico covalent binding, and identified its frontier Molecular Orbitals (MOs) and nucleophilic susceptibility. These physical and chemical reactivity parameters revealed that the bond-forming MOs are distributed on the B ring of the EGCG oxidized state with nucleophilic susceptibility, and that this B ring has properties that favor participating in a Cys-alkylating 1,4-addition reaction. Molecular modeling and docking analysis further revealed that EGCG bonds covalently with Cys-38 of NF-κB-p65, and thereby inhibits its DNA binding ability. We also generated a model pharmacophore based on the EGCG-NF-κB complex. We conclude that EGCG covalently binds to NF-κB-p65 and inhibits it by abolishing its DNA binding, by chemical mechanisms that may inform design of EGCG derivatives as novel anti-inflammatory agents.
Keywords: Epigallocatechin gallate; NF-κB; Covalent bond; 1,4 addition; Cys-alkylation; Molecular modeling; Pharmacophore; Tea
Abbreviations
EGCG: Epigallocatechin Gallate; NF-κB: Nuclear Factor- κB; RHD: Rel Homology Domain; IκBs: Inhibitory κB Proteins; Cys: Cysteine Residue(s); CVDs: Cardiovascular Diseases; MOs: Molecular Orbitals; CΒ: Β-Carbon; DFT: Density Functional Theory; LDA: Local Density Approximation; ADF: Amsterdam Density Functional; QTAIM: Quantum Theory of Atoms in Molecules; PM6: Parameterization Method 6; HOMO: Highest Occupied Molecular orbital; LUMO: Lowest Unoccupied Molecular Orbital; CHARMM: Chemistry at HARvard Macromolecular Mechanics; NP Dock: Nucleic Acid-Protein Docking; QZ4P: Quadruple Zeta with 4 Polarization Functions; H: Hydrogen; HBA: H Bond Acceptors; HBD: H Bond Donors; HY: Hydrophobic; CADD: Computer-Aided Drug Design
Introduction
Cardiovascular Diseases (CVDs) are the leading cause of death worldwide, and will give rise to a predicted increase in annual deaths from 17.5 million in 2012 to 22.2 million by 2030 if current trends persist [1]. CVDs including congestive heart failure, stroke, ischemic and coronary heart disease, coronary artery disease, and peripheral vascular disease [2] inflict high societal costs, and >75% of deaths in countries of low and middle incomes. Worldwide, millions of people strive to control CVD risk factors, while others are unaware of the risks [3]. Inflammation and apoptosis are major pathogenic contributors to these conditions, and induction of many of the pathways involved is heavily mediated by activation of the transcription factor nuclear factor-κB (NF-κB) [4]. CVDs in which NF-κB activation plays an essential pathogenic role include myocardial infarction [5], ischemia/ reperfusion injury [6], transplant rejection [7], angina pectoris [8], autoimmune myocarditis [9], congestive heart failure [10], and cardiomyocyte hypertrophy [11]. Therefore, modulators of NF-κB activity can influence these conditions.
NF-κB consists of a group of structurally-related transcription factors including NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), RelB, and C-Rel, each characterized by a highly conserved Rel homology domain (RHD), the domain which regulates its interaction with inhibitory κB proteins (IκBs), dimerization, and DNA binding to evoke changes in target gene expression [12]. Interactions among NF-κB family members lead to formation of homodimers or heterodimers, among which the most abundant and wellcharacterized is the p50/p65 heterodimer [13]. The p50/p65 dimer interacts with consensus DNA sequences known as κB motifs, which are located in promoter or enhancer regions of target genes, and consist of 5'-GGGRNNYYCC-3', where R is an unspecified purine, Y is an unspecified pyrimidine, and N is any nucleotide [14]. As a result of its activation by cytokines, pathogens, and other stressful conditions, NF-κB induces production of numerous inflammatory mediators including cytokines, chemokines, adhesion molecules, inducible enzymes, and growth factors [15,16]. A highly conserved cysteine residue (Cys-38 in human NF-κB-p65 RHD) is required for its interaction with κB DNA [17]. Several natural and synthetic antioxidant compounds that contain functional electrophilic carbons can inhibit NF-κB DNA-binding activity, by alkylating Cys-38 [18] via a 1,4-addition (S-alkylation) reaction [19].
Antioxidant polyphenolic catechins are present in many nutrientrich foods, such as fruits, berries, and leaves (especially tea), and their health benefits via such antioxidant properties have been wellestablished by in vivo and in vitro studies (reviewed in [20]). Many such beneficial antioxidant activities are attributed to flavonoids that contain dihydroxy or trihydroxy groups, and their antioxidant activity further increases with increasing content of these groups [21-23]. Among the catechins in tea, the most abundant is epigallocatechin gallate (EGCG; C22H18O11) is [24], which can covalently modify proteins and alter their functions [25-27]. Such chemical activities of EGCG reside in its two adjacent trihydroxy structures, the B (gallyl) and D (gallate) rings. These can readily undergo auto-oxidation to form a semiquinone that then rearranges to an electrondeficient and electrophilic Β-carbon (CΒ)- containing O-quinone, which is susceptible to nucleophilic attack by thiols. Such electrophilicnucleophilic attack forms EGCG-S-cysteinyl protein adducts.
EGCG exerts cardiovascular protection via its antioxidant, anti-inflammatory, hypolipidemic, anti-thrombogenic, and antihypertensive actions [28]. Antioxidant properties of catechins include free radical scavenging [29], metal ion chelation [30], inhibition of redox responses, and induction of antioxidant enzymes [31]. EGCG-mediated inhibition of NF-κB via multiple mechanisms [20] contributes to its anti-inflammatory activities. Catechins in tea also improve blood lipid profiles, regulate vascular tone, and impede progression of atherosclerotic lesions, by inhibiting cytokine production, inflammatory cell transmigration, platelet adhesion, and vascular smooth muscle cell proliferation [reviewed in [28]]. Experimental and clinical studies identified protective roles of EGCG in CVDs [32], attracting attention toward developing novel therapeutic strategies targeting Nrf2 activation and NF-kB inhibition [33].
We recently found that EGCG selectively and covalently binds to cysteinyl thiol of NF-κB via 1, 4-addition reaction and effectively suppresses its activation. The cysteine found as the reactive sulfhydryl moiety as S-carboxymethylation blocked the 1, 4-addition reaction between EGCG and NF-κB [34]. Based on our previous findings and EGCG’s biochemical properties, we hypothesized that EGCG covalently binds to NF-κB and inhibits NF-κB-p65’s DNA binding ability. To test this idea we analyzed the operant mechanisms and explored the potential for targeting the relevant sites pharmacologically for therapeutic benefits, by characterizing EGCG’s chemical reactivity and electrophilicity. We found that its oxidized B ring contains proton donating O-quinones and CΒs, which therefore readily undergo chemical reactions. Herein we describe further analyses of the frontier Molecular Orbitals (MOs), nucleophilic susceptibilities, molecular modeling and docking, and identified a new putative pharmacophore based on the EGCG-NF- κB complex. These findings will inform further in silico and in vitro research to enable design of novel EGCG derivatives as potential NF-κB inhibitors.
Materials and Methods
Computational methods
Structures of EGCG in the reduced state (EGCG-RS; C22H18O11) and oxidized state (EGCG-OS; C22H16O11) were generated in ChemOffice (v 17.0, CambridgeSoft, Cambridge, MA, USA). Geometric optimizations and all electronic structure calculations were performed as we described [35], using Density Functional Theory (DFT) by Local Density Approximation (LDA) exchangecorrelation and the QZ4P base set with Amsterdam Density Functional (ADF) Modelling Suite [36]. The critical points, bond paths, atomic properties and energies, and reactivity indices were analyzed via the Quantum Theory of Atoms in Molecules (QTAIM) proposed by Bader, as implemented in ADF. For semi-empirical quantum chemical calculations we used using Parameterization Method 6 (PM6) as implemented by SCiGRESS (v 2.8.1, Fujitsu Ltd., Tokyo, Japan). The resulting parameters, Energies of Highest Occupied Molecular Orbital (EHOMO), and Lowest Unoccupied Molecular Orbital (ELUMO) values were used in standard equations, to determine global chemical reactivity descriptors including hardness (η), chemical softness (σ), chemical potential (μ), electrophilicity (ω), nucleophilicity (ω-), and local reactivity descriptors including the Fukui functions (ƒ+ and ƒ-), Koopmans DD, and philicities.
Molecular modeling
We selected the X-ray structure 1vkx [14] from the RCSB Protein Data Bank to build docking receptors. NF-κB-p65/p50 heterodimer complexed with κB DNA, its C38S mutant (the cysteine at the residue 38 was substituted with a serine), p65 wild-type subunit, and p65 C38S mutant subunit with BIOVIA Discovery Studio (BIOVIA, San Diego, CA, USA). Energy minimization (constraining the heavy atoms) analysis was performed using Chemistry at HARvard Macromolecular Mechanics (CHARMM) force fields [37].
Molecular docking
Molecular docking studies were carried out as we described [38] with Discovery Studio. Briefly, the geometry-optimized EGCG structure, generated based on DFT was used as a ligand. The energyminimized three-dimensional structures and complexes (as described above) were used as receptor molecules to model covalent docking. We further characterized the lowest energy pose of the covalent EGCG-p65 wild-type complex to determine ring conformation changes and generate a pharmacophore model using the receptorligand complex based-common features mapping model.
In vitro electrophilic addition reaction
To determine the EGCG covalent adduction of NF-κB and the involvement of Cys residue an in vitro electrophilic adduction was performed as we described previously [34]. Briefly, unmodified NF- κB-p65 recombinant protein or S-carboxymethylated NF-κB-p65 protein was incubated with various concentrations of biotin taged EGCG. After incubation the formation of the covalent adduction was determined by Western blotting as we previously reported [38].
Nucleic acid–protein docking
We analyzed DNA-protein docking studies as we described [39] to identify interactions between EGCG-bound NF-κB and κB (5′-TGGGGACTTTCC-3′) in the nucleic acid-protein docking (NP Dock) server as described [40], with default docking parameters. We set the RMSD threshold 5 Å for clustering, and used the best-scored decoy in the clusters with the highest probability to identify DNA– protein interactions using Discovery Studio Visualizer.
Results
Calculated chemical properties of EGCG
We determined electrophilicity of EGCG-RS (Figure 1A) and EGCG-OS (Figure 1B) by quantum chemical calculations. We analyzed chemical properties including chemical hardness (η), chemical softness (s), chemical potential (μ), electrophilicity (ω), and nucleophilicity or reactivity (ω-) indices (Tables 1 and 2). The data in Table 1 indicate that EGCG-OS is softer (s: 0.278 eV-1), more reactive (μ: -5.900 eV), and more highly electrophilic (ω: 4.835 eV) than EGCG-G.
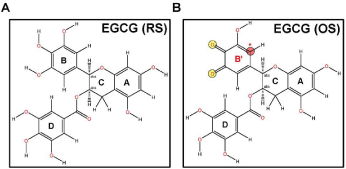
Figure 1: Chemical structures of EGCG. Structures of (A) EGCG-RS
(C22H18O11) and (B) EGCG-OS (B′ ring), drawn using ChemOffice. Figures
were generated with Chem3D. The potential site for 1,4-addition is indicated
by red asterisk (*).
![]()
Molecule
η (eV)
s (eV-1)
μ (eV)
ω (eV)
EGCG (RS)
4.1
0.244
-5.16
3.247
EGCG (OS)
3.6
0.278
-5.9
4.835
Table 1: Calculated quantum mechanical parameters of EGCG. Abbreviations: η, chemical hardness; s, chemical softness; μ, chemical potential; ω, electrophilicity index.
Table 2 shows that the reactivity indices for EGCG-OS with either nucleophile (Cys-S-; thiolate and CysSH; neutral) were higher than that of EGCG-RS, defining the oxidized EGCG, which contains an electrophilic Cβ, as the more likely chemical EGCG form to target biological nucleophiles such as protein thiolates.
![]()
Electrophile
Nucleophile
ω- (eV)
EGCG (RS)
Cys-SH
0.005
Cys-S-
0.0924
EGCG (OS)
Cys-SH
0.045
Cys-S-
0.2156
Table 2: Calculated nucleophilicity (reactivity) index for Cys (biological nucleophile) with EGCG electrophiles. Abbreviation: ω-, reactivity index.
Calculated charge distributions of EGCG
We characterized the charge distribution and bonding nature of the MO by analyzing Mulliken charges derived from the molecules’ Mulliken population. Figure 2 shows the EGCG-RS and EGCGOS atoms, color-coded according to their calculated Mulliken atomic charge populations. Oxidation of O-diphenolic rings in EGCG generates corresponding highly electrophilic O-quinones, with negative charges (bottom colors in Figure 2 Mulliken scales) distributed uniformly across the oxygen atoms of phenolic OH groups (Figure 2). The B ring Oquinones have lower Mulliken charge values than the D ring O-quinones, indicating that the former are potentially proton donors, and preferred, more efficient sites of chemical reactions (Figure 2).
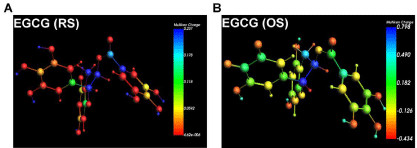
Figure 2: Mulliken atomic charge populations of EGCG. Geometryoptimized
structures of (A) EGCG-RS and (B) EGCG-OS colored according
to Mulliken atomic charges as represented in the color scale. Figures were
generated with ADF.
Frontier Molecular Orbitals (MOs) of EGCG
To determine the electronic structure and MOs, we used Density Functional analyses of EGCG-RS and EGCG-OS, using the QZ4P base set, which is better-polarized than the other basis sets, and encompasses an additional diffuse function. Because Cys-38 in NF- κB-p65 is theoretically a target for 1,4-addition (Michael) reaction, we tested its propensity as such by analyzing electronic structure. In Michael reactions, the electrophilic Cβ forms a covalent bond with a nucleophile, mainly with the thiols of Cysteine Residues (Cys) of cellular peptides and proteins. A covalent bond is formed when the electron-donating HOMO of a nucleophile (Cys Sγ) overlaps with electron-withdrawing LUMO of the electrophile (Cβ) [35, 41]. The distributions of HOMO (Figure 3A; red and blue isosurface of 0.06 au) and LUMO (Figure 3B; orange and cyan isosurface of 0.06 au) we found in EGCG-RS and EGCG-OS indicate that LUMOs are distributed on the B ring in the EGCG-OS containing O-quinones, which act as the electrophilic Cβ. In contrast, in EGCG-RS, we found they are distributed across the C and D rings, indicating the B ring is a potential site of nucleophilic attack. Proton abstraction from the oxygen atoms requires smaller energy than the energy (8.82 eV) required for oxygen ionization. These results indicate that the 1,4-addition reaction of EGCG with Cys-38 of NF-κB-p65 occurs primarily on the B ring.
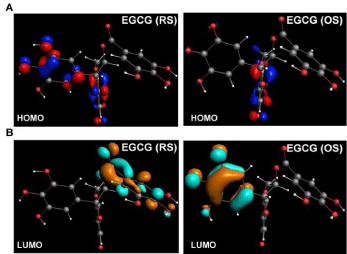
Figure 3: Frontier molecular orbitals (MOs) of EGCG. Geometry-optimized
structures of EGCG-RS and EGCG-OS showing (A) HOMO (red and blue
isosurface; 0.06 au) and (B) LUMO (orange and cyan isosurface; 0.06 au)
distribution. Figures were generated with ADF.
Electrophilicity and Nucleophilic index of EGCG
In addition to identifying chemical descriptors and MOs, we analyzed chemical reactivity of EGCG in terms of electrophilicity and susceptibility (Figure 4). Electrophilicity per atom was determined by condensed Fukui Functions (also called atomic Fukui indices), which quantify the electron density after adding or removing a charge. We set the charge change parameter to 1 in these calculations. For an electrophile, an atom with high Fukui ƒ+ (Fukui function for the nucleophilic attack) value is the one most susceptible to nucleophilic attack.
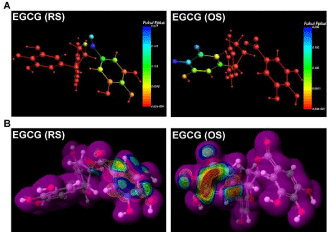
Figure 4: Chemical reactivity of EGCG. Geometry-optimized structures
of EGCG-RS and EGCG-OS showing (A) Fukui ƒ+ distribution colored
to indicate Fukui ƒ+ value, generated with ADF software; and (B) electron
density surface (0.01 au) colored to indicate nucleophilic susceptibility,
generated using SCiGRESS.
As shown in Figure 4A, the highest Fukui values are distributed on the O-quinones of the B ring in EGCG-OS. After determining MOs, we generated a three-dimensional electron density isosurface of EGCG to identify the reactive centers. The van der Waals isosurface of EGCG with relative reactivity is shown in Figure 4B. The surface color indicates susceptibility to nucleophilic attack (with relative reactivity red>blue). These results indicate that the B ring of oxidized EGCG is chemically reactive and is susceptible to nucleophilic attack.
Molecular interactions of the EGCG-NF-κB-p65 complex
To determine the nature of chemical interaction of EGCG with NF-κB-p65, and within that the potential role of NF-κB-p65 Cys-38, we modeled and docked different EGCG-NF-κB complexes. Figure 5 shows the lowest-energy pose of each complex, in which EGCG occupied a groove on the NF-κB-p65 receptor. The complex consists of three hydrogen (H) bonds and various hydrophobic interactions, as shown in 2D-ligand interaction plots. The oxygen atom of the hydroxyl groups in the D ring (galloyl moiety) forms H bonds with the atoms of Tyr-36, Lys-123, and Arg-124. Electrostatic interactions also contribute to EGCG’s binding. For example, electrondense B and D rings interact with positively-charged amino acids such as Lys-122. The nitrogen atom of the Lys-122 side chain is closely positioned to the oxygen atom of a D ring. EGCG also interacts with several nearby amino acids via van der Waals and hydrophobic interactions, shown in Figure 5C-5D.
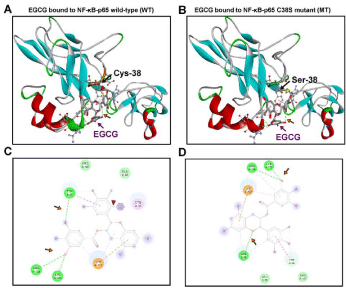
Figure 5: Molecular modeling and docking of the EGCG-NF-κB-p65
complex. Molecular models of EGCG bound to (A) NF-κB-p65 wild-type
subunit and (B) NF-κB-p65 C38S mutant subunit, with respective (C and
D) 2D-ligand interaction plots of the binding site containing EGCG and the
NF-κB-p65 amino acid residues which interact and stabilize the ligand in the
selected docking pose. NF-κB-p65 shown as a secondary structure colored
ribbon. EGCG is shown as an element-colored stick model with interacting
amino acids represented as balls and sticks. The covalent bond between
EGCG and NF-κB-Cys-38 (Cys Sγ interaction with the electrophilic Cβ of
EGCG) is indicated by red arrowhead (A and C); its absence is indicated
by yellow arrowhead (B). H bonds in green dotted lines are indicated with
an orange arrow. Figures were generated with Discovery Studio Visualizer.
EGCG only formed a covalent bond with the wild-type NF- κB-p65 complex (Figure 5A and 5C.; indicated by red arrowhead), and covalent interaction was abolished by C38S mutation in the NF- κB-p65 C38S mutant complex (Figure 5B and 5D). In the covalent conformation (Figure 5A and 5C), the electrophilic Cβ in the EGCG’s B ring was separated by ~2 Å from the sulfur atom (Sγ) of Cys38, a distance favorable for a 1,4-addition reaction. These results indicate that EGCG covalently binds to NF-κB-p65, via Cys-38.
EGCG-NF-κB-p65 form a Cys covalent adduct
We next tested the covalent adduction between EGCG and NF-κB-p65 by performing an electrophilic addition reaction assay. We found that EGCG covalently and dose-dependently interacts with NF-κB-p65 and the interaction was competed by unlabeled EGCG. Additionally, the interaction was abolished in the presence of diamide an oxidizing agent, demonstrating the covalent nature of the interaction (Figure 6A). As molecular docking studies showed the necessity of Cys-38 for the covalent interactions, we tested the involvement of Cys residues by blocking their adduction by S-carboxymethylation. As predicted, blocking Cys residues abolished the covalent adduction demonstrating their involvement and necessity (Figure 6B). These experimental observations prove the in silico molecular findings.
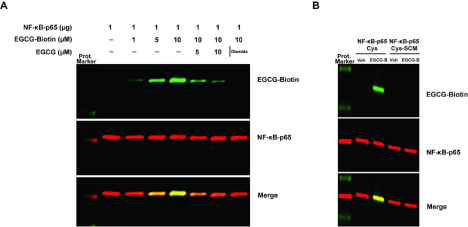
Figure 6: EGCG and NF-κB-p65 covalent adduction. (A) NF-κB-p65 (1 μg) protein was incubated with various concentrations of EGCG-Biotin, unlabeled EGCG
and diamide. (B) NF-κB-p65Cys or NF-κB-p65-Cys-SCM (S-carboxymethylated) protein was incubated with EGCG-Biotin. Following incubation the covalent
adduction was determined by SDS-PAGE and Western blotting.
EGCG-NF-κB-p65 pharmacophore
Receptor-ligand complex-based pharmacophore generation identifies the chemical features which initiate the key interactions between a ligand and target receptor. We generated a pharmacophore based on the EGCG-NF-κB-p65 covalent complex (Figure 7). The pharmacophore mapped all common features of the interactions between EGCG and NF-κB-p65, which contain two H Bond Acceptors (HBA) and five H Bond Donors (HBD) (Figure 7A). EGCG contains four main pharmacophoric features (Figure 7B), including one HBA, two HBD, two Ring-Aromatic (RA), and one Hydrophobic (HY) feature. The distance constraints between the inter-chemical features of EGCG-NF-κB-p65 complex reveal the critical positioning of the chemical features of EGCG (Figure 7C). These results validate the required chemical features of EGCG for its binding with target proteins. Further optimization in future may guide design of novel EGCG derivatives as potential NF-κB inhibitors.

Figure 7: Pharmacophore of the EGCG-NF-κB-p65 complex. (A-C) Pharmacophore mapping of EGCG-NF-κB-p65 covalent docking model, showing the H
bond donors (HBDs; magenta), the H bond acceptors (HBAs; green), the ring aromatics (RAs; orange), and the hydrophobic features and locations (HY; cyan).
EGCG represented as a line model with a HBD-HBA surface. 3D spatial arrangement and distance constraints between chemical features of the EGCG-NF-κB-p65
complex are shown in (C). Figures were generated with Discovery Studio Visualizer.
EGCG is an NF-κB-p65 DNA binding inhibitor
The Rel Homology Domain (RHD) of NF-κB contains a highlyconserved cysteine (Cys-38), a key residue that facilitates NF-κB binding to DNA [42]. Cys-38 interacts with the phosphate backbone of the κB DNA enhancer motif, and alkylation of Cys-38 inhibits DNA binding activity of NF-κB [17]. As our results showed that EGCG binds covalently to the Cys-38 residue of NFκB-p65 via 1,4-additions (S-alkylation), we analyzed the effect of NF-κB-p65’s binding of EGCG on its DNA-binding activity. As shown in Figure 8A, NF-κB-p65 binds to the κB DNA enhancer motif, and Cys-38 (acting as a HBD) formed a conventional H bond with the oxygen atom in the phosphate of thymidine, at a distance of 2.76 Å (indicated by green arrow). However, the EGCGbound NF-κB-p65 failed to interact with the κB DNA enhancer motif (as shown in Figure 8B). Covalent binding of EGCG (indicated by red arrowhead) positioned Cys-38 away from the DNA, and abolished formation of the relevant H bond. The absence of NF-κB-DNA interaction is indicated by the pink arrow. Our results explain the importance of the NF-κB-Cys-38 residue, and role of its covalent modification by antioxidant molecules such as EGCG in inhibiting NF-κB signaling, identifying EGCG as a novel inhibitor of NF-κB-p65 binding to its cognate motif in DNA.
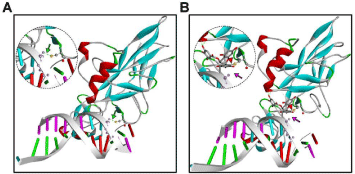
Figure 8: DNA interactions of NF-κB-p65. Molecular models of DNA (κB; 5′-TGGGGACTTTCC3′) interactions of (A) NF-κB-p65 (B) EGCG-bound NF-κB-p65. NF-κB-p65 shown as a secondary structure colored ribbon. EGCG is shown as an element-colored stick model with interacting amino acids represented as balls and sticks. Red arrowhead indicates the covalent bond between EGCG and NF-κB-Cys-38. Presence of H bond interactions between κB and NF-κB-p65 (Cys Sγ [yellow]) interaction with the phosphate backbone of κB DNA) is indicated by green arrow, and its absence by pink arrow. Inserts show close-up views of the interactions. Figures were generated with Discovery Studio Visualizer.
Discussion
In modern drug discovery, theoretical and computational approaches such as ligand-, receptor-, and pharmacophore-based methods (Computer-Aided Drug Design [CADD]) inform design of novel and potential drugs. Drugs developed by CADD evolve by additional experimental studies, and have developed into drugs in clinical use [43]. Natural products provide an enormous advantage for ligand- and pharmacophore-based drug design, to develop standalone and structural (synthetic) derivative drugs [44]. Tea is such a natural dietary product, that has been extensively used in traditional medicine [45], and EGCG, tea’s most abundant antioxidant catechin, is likely a key contributor of tea’s health benefits [21].
EGCG interferes with many disease-related signaling mechanisms, specifically by inhibiting a small number of targets, at biologically relevant concentrations delivered by dietary consumption [46]. Its beneficial effects in CVDs have been extensively tested experimentally and in prospective cohort studies [30, 47]. Our present findings reveal molecular details of the mechanism of EGCG-mediated inhibition of NF-κB. Prior in silico studies analyzed EGCG’s antioxidant activity [48] and interactions with proteasomes [49], human serum albumin [50], trypsin [51], and B-Raf [52], but the precise chemical mechanisms of its inhibition of NF-κB was unknown. We recently identified EGCG as a novel covalent NF-κB inhibitor [34], and here we revealed key parameters of its chemical reactivity including electrophilic Fukui ƒ+ function, in silico covalent binding, and generated the first known EGCG-NF-κB complex-based pharmacophore. The quantum mechanical parameters we computed revealed that EGCG-OS has electrophilicity index of 4.835 eV and reactivity index of 0.2156 eV with cysteine thiolate. Our density functional and chemical reactivity calculations showed that the B ring of EGCG-OS has properties that confer potential to participate in the Cys-alkylating 1,4-addition reaction. By molecular docking analyses, we found that EGCG covalently binds to NF-κB-p65 and abolishes its DNA binding. By generating the indicated pharmacophore, we further validated the importance of the required chemical features to EGCG’s complexation with NF-κB.
Owing to the crucial roles of NF-κB and the pathways that control its activation in human disorders such as chronic inflammatory diseases and CVDs, translational/clinical studies have assessed various compounds’ abilities to inhibit NF-κB activity and their mechanisms of action. Many of these exhibit cross-reactivity with other important signaling pathways such as p53 [53], as reviewed [54]. Among these, inhibitors that target NF-κB by direct covalent modification of redoxregulated cysteine residues in NF-κB subunits have attracted interest [18] as potentially useful inhibitors of NF-κB activation, via upstream blockade of IKKβ activation and downstream blocking of dimeric NF-κB DNA-binding.
Pharmacological agents which can modulate NF-κB activation at different stages may prove useful in clinical applications or research, as a wide range of distinct stimulus types activate NF-κB, and these depend to some extent upon on cell type [55]. Development of such inhibitors may thus facilitate further translational/clinical research on NF-κB inhibitors as potential therapeutic agents, targeting chronic inflammatory diseases such as CVDs. We are extended our hypothesis and findings in in vitro studies to identify covalent modification of NF-κB by EGCG, the involvement of specific Cys residues, yet the biological and physiological significance of this covalent interaction need to be understood. Further optimizing the identified EGCG-NF-κB-p65 pharmacophore will inform and enable design of new and biologically or clinically useful NF-κB inhibitors, and ultimately the preclinical research and any human trials needed to elucidate the molecular and health-beneficial effects of antioxidant polyphenolic catechins in humans, and recommend dosing and effective applications in disease conditions including CVDs.
Highlights
• EGCG is a novel covalent inhibitor of NF-κB.
• EGCG’s B ring participates in a Cys-alkylating 1,4-addition reaction.
• Optimizing EGCG-NF-κB-p65 pharmacophore enable design of new and biologically or clinically useful NF-κB inhibitors.
Data Availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Ethical Approval
Not applicable. This article does not directly involve the operation of patients or animals but focuses on the molecular mechanism.
Author Contribution
A.T.R., S.P.L., N.Ch.V., and L.D.K. conceived and designed the experiments; A.T.R. and S.P.L. performed the experiments and analyzed the data; A.T.R., S.P.L., N.Ch.V., and L.D.K. wrote the manuscript.
References
- Cardiovascular diseases (CVDs): World Health Organization (WHO). 2017.
- Luepker RV. Cardiovascular disease: rise, fall, and future prospects. Annual review of public health. 2011; 32: 1-3.
- Hearts: technical package for cardiovascular disease management in primary health care: World Health Organization (WHO). 2016.
- Jones WK, Brown M, Wilhide M, He S, Ren X. NF-κB in cardiovascular disease. Cardiovascular toxicology. 2005; 5: 183-201.
- Sanganalmath S, Cheng G, Girgis M, Xuan Y-T, Yang Y, Elias H, et al. Cardiacspecific transgenic silencing of NF-κB ameliorates left ventricular dysfunction and remodeling after acute myocardial infarction. Journal of the American College of Cardiology. 2012; 59: E909.
- Maimaitiaili A, Li J, Aibibula A, Abudureheman M. Inhibition of nuclear factor kappa B pathway protects myocardial ischemia/reperfusion injury in rats under treatment with abnormal savda munziq. American journal of translational research. 2018; 10: 77.
- Tsoulfas G, Geller DA. NF-κB in transplantation: friend or foe? Transplant Infectious Disease: Basic Science. 2001; 3: 212-219.
- Ritchie ME. Nuclear factor-κB is selectively and markedly activated in humans with unstable angina pectoris. Circulation. 1998; 98: 1707-1713.
- Valaperti A. Drugs targeting the canonical NF-κB pathway to treat viral and autoimmune myocarditis. Current pharmaceutical design. 2016; 22: 440-449.
- Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circulation research. 2011; 108: 1122-1132.
- Purcell NH, Molkentin JD. Is nuclear factor κB an attractive therapeutic target for treating cardiac hypertrophy? : Am Heart Assoc. 2003.
- Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annual review of immunology. 1998; 16: 225-260.
- Schmitz ML, Baeuerle PA. The P65 Subunit Is Responsible for the Strong Transcription Activating Potential of Nf-Kappa-B. Embo J. 1991; 10): 3805- 3817.
- Chen FE, Huang D-B, Chen Y-Q, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature. 1998; 391: 410-413.
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009; 1: a000034.
- Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annual review of biophysics. 2013; 42: 443-468.
- Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, et al. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. The Journal of biological chemistry. 2001; 276: 39713-39720.
- Pande V, Sousa S, Ramos M. Direct covalent modification as a strategy to inhibit nuclear factor-kappa B. Current medicinal chemistry. 2009; 16: 4261- 4273.
- Na HK, Surh YJ. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Molecular Carcinogenesis: Published in cooperation with the University of Texas MD Anderson Cancer Center. 2006; 45: 368-380.
- Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin- 3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011; 82: 1807-1821.
- Jovanovic SV, Steenken S, Hara Y, Simic MG. Reduction potentials of flavonoid and model phenoxyl radicals. Which ring in flavonoids is responsible for antioxidant activity? Journal of the Chemical Society, Perkin Transactions 2. 1996; 11: 2497-504.
- Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free radical biology and medicine. 1996; 20: 933-956.
- Silva MM, Santos MR, Caroço G, Rocha R, Justino G, Mira L. Structureantioxidant activity relationships of flavonoids: a re-examination. Free Radical Research. 2002; 36: 1219-1227.
- Clement Y. Can green tea do that? A literature review of the clinical evidence. Preventive medicine. 2009; 49: 83-87.
- Ishii T, Mori T, Tanaka T, Mizuno D, Yamaji R, Kumazawa S, et al. Covalent modification of proteins by green tea polyphenol (-)-epigallocatechin-3-gallate through autoxidation. Free radical biology & medicine. 2008; 45: 1384-1394.
- Sang S, Lambert JD, Hong J, Tian S, Lee MJ, Stark RE, et al. Synthesis and structure identification of thiol conjugates of (-)-epigallocatechin gallate and their urinary levels in mice. Chemical research in toxicology. 2005; 18: 1762-1769.
- Chen R, Wang J-B, Zhang X-Q, Ren J, Zeng C-M. Green tea polyphenol epigallocatechin-3gallate (EGCG) induced intermolecular cross-linking of membrane proteins. Archives of biochemistry and biophysics. 2011; 507: 343-349.
- Babu A, Pon V, Liu D. Green tea catechins and cardiovascular health: an update. Current medicinal chemistry. 2008; 15: 1840-1850.
- Frei B, Higdon JV. Antioxidant activity of tea polyphenols in vivo: evidence from animal studies. The Journal of nutrition. 2003; 133: 3275S-3284S.
- Stangl V, Dreger H, Stangl K, Lorenz M. Molecular targets of tea polyphenols in the cardiovascular system. Cardiovascular research. 2007; 73: 348-358.
- Cabrera C, Artacho R, Gimenez R. Beneficial effects of green tea--a review. J Am Coll Nutr. 2006; 25: 79-99.
- Peters U, Poole C, Arab L. Does tea affect cardiovascular disease? A metaanalysis. American Journal of Epidemiology. 2001; 154: 495-503.
- Fang J, Sureda A, Silva AS, Khan F, Xu S, Nabavi SM. Trends of tea in cardiovascular health and disease: A critical review. Trends in Food Science & Technology. 2019.
- Lakshmi SP, Reddy AT, Kodidhela LD, Varadacharyulu NC. The tea catechin epigallocatechin gallate inhibits NF-κB-mediated transcriptional activation by covalent modification. Archives of Biochemistry and Biophysics. 2020; 695: 108620.
- Reddy AT, Lakshmi SP, Banno A, Reddy RC. Identification and Molecular Characterization of Peroxisome Proliferator-Activated Receptor d as a Novel Target for Covalent Modification by 15-Deoxy-Δ12,14 prostaglandin J2. ACS chemical biology. 2018; 13: 3269-3278.
- Baerends EJ, Ziegler T, Atkins AJ, Autschbach J, Bashford D, Baseggio O, et al. ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam.
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Journal of chemical theory and computation. 2016; 12: 405-413.
- Lakshmi SP, Reddy AT, Banno A, Reddy RC. Molecular, chemical, and structural characterization of prostaglandin A2 as a novel agonist for Nur77. Biochemical Journal. 2019; 476: 2757-2767.
- Lakshmi SP, Reddy AT, Banno A, Reddy RC. Airway Epithelial Cell Peroxisome Proliferator–Activated Receptor γ Regulates Inflammation and Mucin Expression in Allergic Airway Disease. The Journal of Immunology. 2018; 201: 1775-1783.
- Tuszynska I, Magnus M, Jonak K, Dawson W, Bujnicki JM. NPDock: a web server for protein–nucleic acid docking. Nucleic acids research. 2015; 43: W425-W30.
- Poon T, Mundy BP, Shattuck TW. The Michael reaction. J Chem Educ. 2002; 79: 264-267.
- Perkins ND. Cysteine 38 holds the key to NF-kappaB activation. Mol Cell. 2012; 45: 1-3.
- Prieto-Martínez FD, López-López E, Juárez-Mercado KE, Medina-Franco JL. Computational Drug Design Methods—Current and Future Perspectives. In Silico Drug Design: Elsevier. 2019; 19-44.
- Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nature reviews drug discovery. 2015; 14: 111-129.
- Tauber AL, Schweiker SS, Levonis SM. From tea to treatment; epigallocatechin gallate and its potential involvement in minimizing the metabolic changes in cancer. Nutr Res. 2019; 74: 23-36.
- Nagle DG, Ferreira D, Zhou YD. Epigallocatechin-3-gallate (EGCG): chemical and biomedical perspectives. Phytochemistry. 2006; 67: 1849-1855.
- Eng QY, Thanikachalam PV, Ramamurthy S. Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases. Journal of ethnopharmacology. 2018; 210: 296-310.
- Meng Q, Yang Z, Jie G, Gao Y, Zhang X, Li W, et al. Evaluation of antioxidant activity of tea polyphenols by a quantum chemistry calculation method-PM6. Journal of Food and Nutrition Research. 2014; 2: 965-972.
- Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Docking studies and model development of tea polyphenol proteasome inhibitors: applications to rational drug design. Proteins. 2004; 54: 58-70.
- Maiti TK, Ghosh KS, Dasgupta S. Interaction of (-)-epigallocatechin-3-gallate with human serum albumin: fluorescence, fourier transform infrared, circular dichroism, and docking studies. Proteins. 2006; 64: 355-362.
- Cui F, Yang K, Li Y. Investigate the binding of catechins to trypsin using docking and molecular dynamics simulation. Plos One. 2015; 10: e0125848.
- Ying HZ, Xie JF, Liu XG, Yao TT, Dong XW, Hu CQ. Discriminatory analysis based molecular docking study for in silico identification of epigallocatechin- 3-gallate (EGCG) derivatives as B-Raf(V600E) inhibitors. Rsc Adv. 2017; 7: 44820-44826.
- Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-κB pathways. Nature reviews Drug discovery. 2008; 7: 1031-1040.
- Gilmore T, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006; 25: 6887-6899.
- Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nature reviews Molecular cell biology. 2004; 5: 392-401.