
Review Article
Austin J Cerebrovasc Dis & Stroke. 2014;1(4): 1018.
Revisiting Electrophysiological Mechanisms of VF/VT Arrest During Early Ischemia and Spontaneous Electrical Activity After Defibrillation: From Cell to ACLS
Muramatsu H1* and Takayama M2
1Department of Medicine, Nippon Medical School, Japan
2Department of Cardiology, Sakakibara Heart Institute, Japan
*Corresponding author: Muramatsu H, Department of Medicine, Division of Cardiology, Nippon Medical School 7-3 Daiwa, Koufu, Yamanashi 400-0072, Japan
Received: July 25, 2014; Accepted: August 20, 2014; Published: August 22, 2014
Abstract
K+ conductance and [K+]o increase during early (<10 min) regional and global ischemia. Early ischemia depolarizes the RMP and decreases INa (that is, residual INa) causing slow conduction, and shortens APD and ERP with dispersion. All of these factors contribute to the reentry mechanisms of VF/VT. While IK, ATP has a pivotal role in the increase in [K+]o, other currents such as IK, Na, IK, Ca, IK, FAA have simultaneously important effects on the increase in [K+]o. Up regulation of IK1, with an increase in inward-rectification, can also contribute to the increase in [K+]o in very early ischemia. VT is induced by ordered reentry of spiral waves, whereas VF is caused by random reentry of the spiral wavelets’ breakup. A biphasic waveform is more effective to defibrillate VF than a monophonic waveform, because, in the biphasic waveform the first hyper polarization resets every state of Na+ channels to prepare for reopening, and the subsequent depolarization simultaneously and uniformly inactivates almost all of the Na+ channels. Pacemaker restoration with spontaneous electrical activity can originate from either the SAN, AVN or Purkinje cells. Ischemia suppresses pacemaker activity, because pacemaker currents are sensitive to the ischemia, especially in the SAN, which has the fastest firing rate. Post ischemic early reperfusion injury induces a stunned myocardium and contractile disturbance, which can cause post-defibrillation pseudo PEA. After at least 5 min of an induced VF arrest, the reduction of PO2 was statistically significant, but that change was not remarkable; therefore, the estimated SaO2 does not decrease remarkably. High-quality CPR prevents global ischemia of the heart and brain due to VF/pulse less VT arrest. Therefore, it is essential to restore the organized electrical activity of pace making, to facilitate effective contraction of the ventricular muscle (that is, ROSC), and to minimize ischemic and post ischemic injury of these important organs.
Keywords: Acute coronary syndromes; Ventricular fibrillation; Cardiopulmonary and cerebral resuscitation; Defibrillation; Pacemaker cells; Ion channels
Abbreviations
ACLS: Advanced Cardiopulmonary And Cerebral Life Supports; ACS: Acute Coronary Syndromes; ADP: Adenosine Di phosphate; aK o: Extracellular K+ Activity; AMI: Acute Myocardial Infarction; AP: Action Potential; APD: Action Potential Duration; ATP: Adenosine Triphosphate; [ATP]i: Intracellular ATP Concentration; AVN: Atrioventricular Node; BLS: Basic Life Supports; [Ca2+]i: Intracellular Ca2+Concentration; c AMP: Cyclic Adenosine Mono phosphate; CPR: Cardiopulmonary and Cerebral Resuscitation; Cx: Connexins; DAD: Delayed After depolarization; ECG: Electrocardiogram; ERP: Effective Refractory Period; [H+]i: Intracellular H+ Concentration; Ib: Background Current; ICa: Calcium Current; ICaT: T-type Calcium Current; ICaL: L-type Calcium Current; Ih(f): Hyper polarization- Activated (funny) current; IK1: Inwardly Rectifying K+ current; IK: ATP: ATP-Sensitive K+ Current; IK: Ca: Ca2+-Dependent K+ Current; IK: FAA: Fatty Acid-Activated K+ Current; IK: Na: Na+-Activated K+ Current; INa: Na+ Current; INa/Ca: Na+-Ca2+ Exchange Current; INa/K: Na+-K+ Pump Current; IKr: Rapid Component Of Delayed Rectifier Outward K+ Current; IKs: Slow Component Of Delayed Rectifier Outward K+> Current; INSC: Nonselective Cation Channel Current; Ist: Sustained Inward Current; Kd: Equilibrium Dissociation Constant; [K+]e: [K+] o: Extracellular K+ Concentration; LAD: Left Anterior Descending Artery; LCFA: Long Chain Fatty Acid; NA: Noradrenalin; [Na+]i: Intracellular Na+ Concentration; NADPH: Nicotinamide AdenineDinucleotide Phosphate; NBF: Nucleotide Binding Folds; NO: Nitric Oxide; NSTE ACS: Non St-Elevated Acute Coronary Syndromes; NSTEMI: Non St-Elevated Myocardial Infarction; NSVT: Non sustained Ventricular Tachycardia; PCO2: Partial Pressure Of Carbon Dioxide; PEA: Pulse less Electrical Activity; [pH]o: Extracellular Ph; PO2: Partial Pressure Of Oxygen; Q10: Temperature Coefficient; RMP:Resting Membrane Potential; ROS: Reactive Oxygen Species; ROSC: Return Of Spontaneous Circulation; SAN: Sinoatrial Node; SaO2: Saturation of Arterial Oxygen; SCD: Sudden Cardiac Death; SR: Sarcoplasmic Reticulum; STEMI: ST-Elevated Myocardial Infarction; SUR: Sulfonylurea Receptor; UA: Unstable Angina; VF: Ventricular Fibrillation; VT: Ventricular Tachycardia.
Introduction
Patients with coronary atherosclerosis may develop a spectrum of clinical syndromes representing varying degrees of coronary artery occlusion. Acute coronary syndromes (ACS) include ST-elevated myocardial infarction (STEMI) and non ST-elevated ACS. The latter includes unstable angina (UA) and non ST-elevated myocardial infarction (NSTEMI) due to incomplete occlusion of the coronary artery: whereas the former is due to complete occlusion. Sudden cardiac death (SCD) may occur with each of these syndromes [1,2].
Approximately 52% of acute myocardial infarction (AMI) manifests as the SCD that relates to ventricular fibrillation (VF): and occurs mostly within the first hours after the onset of symptoms: inevitably causing prehospital deaths [1,2]. Furthermore: in about 30% of cases: lethal ventricular arrhythmias appear within 30 min from the onset of the AMI: leading to SCD without appropriate emergency cardiovascular care [3].
Basic life supports (BLS) and advanced cardiopulmonary and cerebral life supports (ACLS) are essential to emergency cardiovascular care. The chain of survival is a critical concept to perform resuscitation: including in adult patients (1) immediate recognition of cardiac arrest and activation of the emergency response system; (2) early cardiopulmonary and cerebral resuscitation (CPR) with an emphasis on chest compressions; (3) rapid defibrillation; (4) effective ACLS; (5) integrated immediate post-cardiac arrest care; and (6) rehabilitation [1,2]. The rehabilitation care for patients who survive SCD requires comprehensive cardiac and cerebral rehabilitation.
However: in order for the surviving patients to have a good prognosis: a critical component in both BLS and ACLS is continuous high-quality CPR in which the chest compressions are especially important [1]. Continuous high-quality CPR: early after arrest from VF or pulse less ventricular tachycardia (VT) and immediately after electrical defibrillation: maintains cerebral as well as coronary blood perfusions. High-quality CPR also can suppress progressive global ischemia in the whole heart during VF/VT arrest: decrease arrhythmogenic substrates: wash out ischemia-related injurious products: and provide oxygen and glucose for pacemaker cells to generate spontaneous electrical activity and for ventricular cells to contract.
Thus: understanding of the cellular pathophysiological and electrophysiological mechanisms that underscore the need for the continuous high-quality CPR and early defibrillation: besides simplified practical algorithms: could be helpful for BLS and ACLS education: training and clinical practice.
Understanding the electrophysiological mechanisms is crucial: especially for the following.
- Malignant ventricular arrhythmias: such as VT and/or VF: during early ischemia: i.e. a sudden cardiac arrest in the ACS.
- More beneficial effect of a biphasic waveform to electrically defibrillate the ventricles.
- Spontaneous electrical activity of pacemaker cells and ventricular contraction after successful defibrillation: which assure a return of spontaneous circulation (ROSC).
- Post-defibrillation pseudo pulse less electrical activity (PEA) associated with reperfusion contractile disturbance: i.e. ““stunned myocardium”: after the global ischemia due to the VF/VT arrest.
Although many resources now are available: a basic knowledge of the electrophysiological mechanisms during resuscitation: particularly ion channels and cellular metabolism: is quite limited. Therefore: here we briefly review and summarize what is known about those mechanisms: while emphasizing the need for high-quality CPR during early VF/VT arrest from ACS.
Mechanisms of VF/VT in early ischemia
The triphasic time course of extracellular K+ accumulation in acute regional myocardial ischemia
As is generally accepted: Figure 1 shows that increases in extracellular K+ concentration ([K+]o) and extracellular K+ activity (aKo) occur in a triphasic pattern: that is: phases I : II and III [4]. In phase I: the initial rapid phase of [K+]o accumulation or aKo increase began to rise within 15-20 sec. In phase II: a plateau occurred after approximately 10-15 min of ischemia: ranging 10.7-18 mM [K+] o or 8-13 mM aKo. The dotted line indicates an occasional minor decline that may appear in some experiments. In phase III: a second progressive increase was no longer reversible upon reperfusion [4]. The slope of phase I and the time and level of phase II varied with different experimental conditions and species.
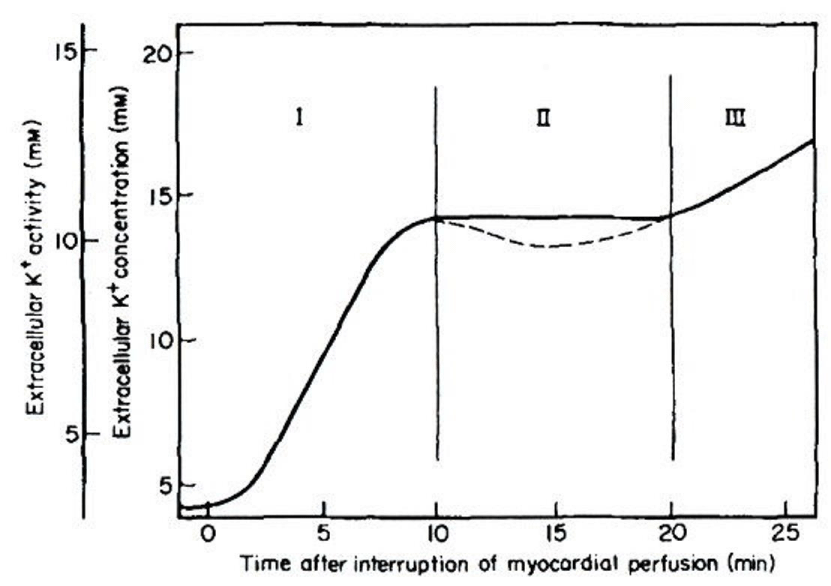
Figure 1 : Increase in [K+]o by coronary artery ligation in Langendorff-perfused mammalian hearts. Modified from [4], with permission of Elsevier.
Malignant ventricular arrhythmias in Harris’ phase 1 correspond with the increase in [K+]o during phases I and III
In Figure 2: a regional ischemia was produced by ligation of the left anterior descending coronary artery (LAD) in swine hearts [5]. In Harris’ experiments: malignant ventricular arrhythmias occurred in two distinct phases that were separated by a period of relative quiescence for approximately 10 min [5]. In phase 1a: the first burst of VT was seen between 2 and 10 min: during which evolution into VF can be rare (~10%). In phase 1b: the second phase of the malignant ventricular arrhythmias appeared between ~20 and 35 min: during which deterioration to VF and death is more frequent (~70%). A continuous increase in tissue resistance: as a result of the more elevated [K+]o and lower [pH]o: is accompanied by a greater probability of VF [5,6].
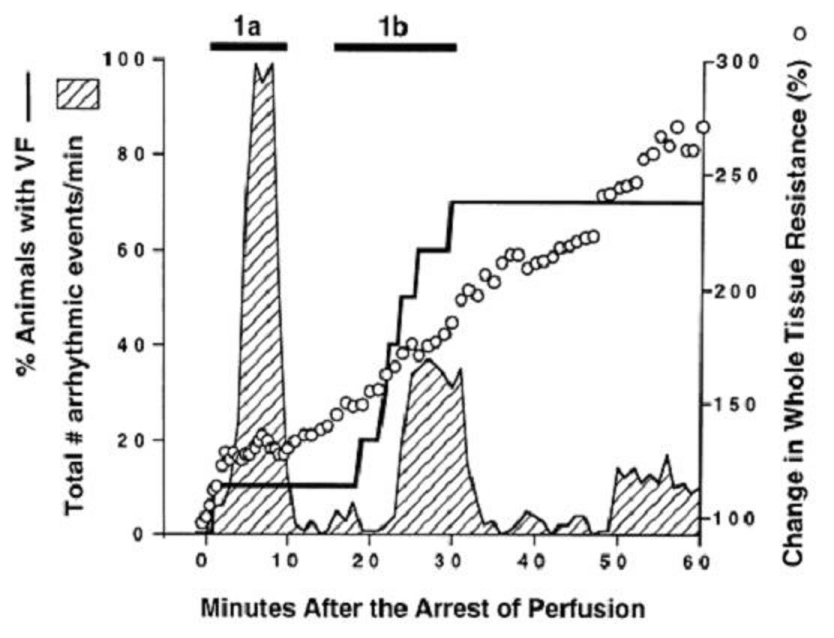
Figure 2 : Two phases (1a, 1b) of malignant ventricular arrhythmias can be distinguished during acute cardiac regional ischemia. Modified from [5], with permission of the American Association for the Advancement of Science.
Note that Harris’ phase 2 and 3 arrhythmias occurred later: not during the very early ischemia: in different periods and by different mechanisms [5]. Phase 2 spanned from 3-6 hr to 24-72 hr. numerous ventricular ectopic beats and short salvos of VT originated from the subendocardial Purkinje fibers due to abnormal automaticity that was precipitated by excessive sympathetic tone: rather than triggered activity or reentry. In phase 3: after more than 5 days following the infarction: premature ventricular beats and short runs of VT occurred due to reentry originating from the surviving cells overlying the infarct or from the border zone.
Underlying K+ currents responsible for the early increase in [K+]o
In Figure 3: acute “regional” ischemia was produced by coronary ligation of Langendorff-perfused quiescent and 4 Hz-stimulated (240 bpm) rat hearts [7]. K+-sensitive electrodes: inserted in the mid-left myocardium: were employed to measure [K+]e. The phase I increase in [K+]e appeared more rapidly in stimulated hearts than in quiescent hearts. The phase II plateau was absent from the quiescent hearts [7]. Because adenosine triphosphate (ATP) was still maintained in phase II: between ~5 and 12 min: the presence of a plateau was interpreted to arise from accelerated electrogenic Na+/K+ pump current (INa/K) to remove the increased Na+ from ischemic myocardial cells. In Figure 4: K+-sensitive electrodes measured [K+]o in “globally” ischemic rabbit hearts stimulated at 3.5 Hz (210 bpm) [8]. Although glibenclamide (3 μM) totally blocks the ATP-sensitive K+ current (IK: ATP): it only partially (~50%) attenuated the phase I increase in [K+]o. It should be noted that the sub sarcolemmal concentration of ATP can be maintained at least up to phase II: during which INa/K is quite activated with the preserved Na+/K+ ATPase activity. Furthermore: a nuclear magnetic resonance study has revealed that bulk intracellular ATP concentration ([ATP]i) is only slightly reduced (~ 2 mM) during very early (≤ 10 min) ischemia [9]. Thus: although IK: ATP is a pivotal player during very early (phase I) ischemia: there are other factors as well.
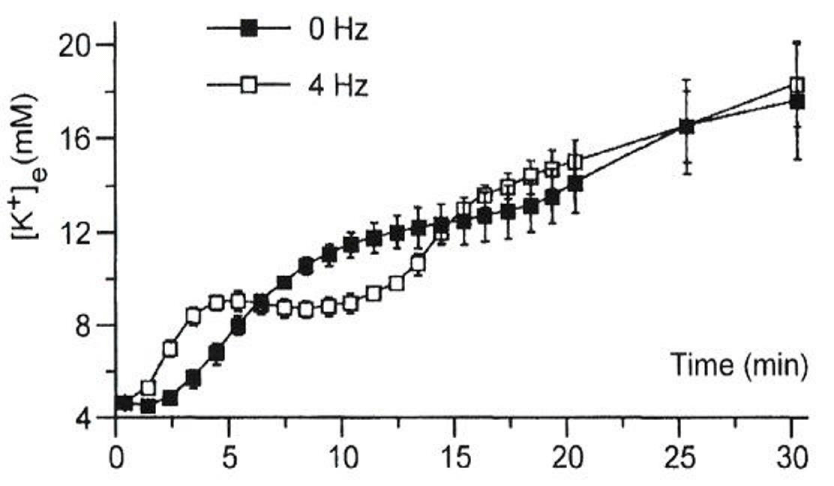
Figure 3 : Extracellular K+ accumulation during very early ischemia. Modified from [7], with permission of Oxford University Press.
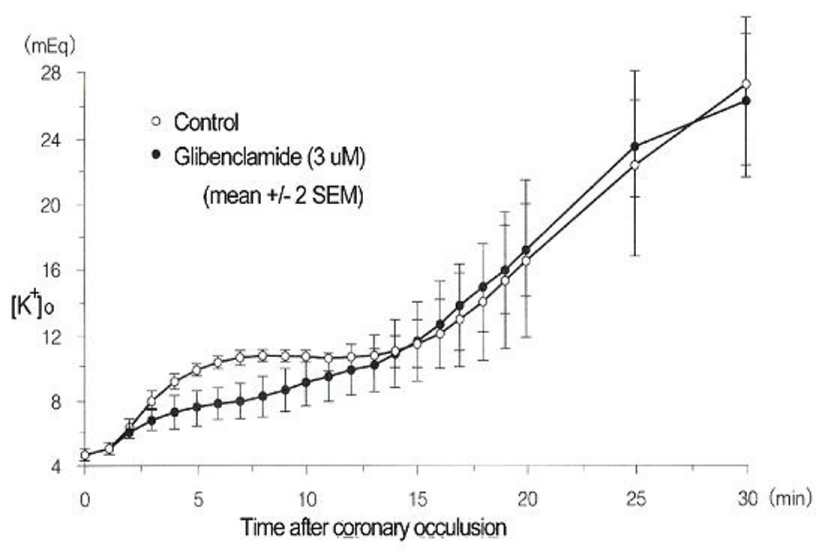
Figure 4 : A role of the ATP-sensitive K+ channel in the early (phase I) K+- efflux and increase in [K+]o. Modified from [8], with permission of Wolters Kluwer Health.
Activation of the ATP-sensitive K+ channel and contributing factors to the channel
In general: KATP channels open when [ATP]i falls from ≥ 5 mM: in normal conditions: to below 1-2 mM. Especially in inside-out membrane patch experiments: the Kd for ATP is only ~ 0.1 mM: which is far less than the normal value [10]. In fact: [ATP]i decreases only a little (~ 2 mM) during early ischemia (phase I-II) [9]. Still: a pivotal role of IK: ATP in the increase in [K+]o during phase I cannot be ignored. Some reasonable explanations for this discrepancy are the following. Sub sarcolemmal [ATP]i: where the Na+/K+ ATPase or adenyl cyclase is activated (“ATP compartmentalization”): is much lower than the bulk [ATP]i: [11,12].
ATP inhibition of KATP channels is relieved by local accumulation of adenosine diphosphate (ADP): adenosine and lactate: which are generated by ATP degradation and anaerobic glycolysis [13,14].
The KATP channel functions as a hetero-octamer: which is assembled from four Kir 6.2 subunits (tetramer) and four sulfonylurea receptor (SUR) 2A subunits (tetramer)? MgADP: binding to nucleotide binding folds (NBF)-2: antagonizes the ATP binding at NBF-1 of SUR2A with high affinity. Thus: an increase in the ATP/Mg ADP ratio closes KATP channels: whereas a decrease in the ratio readily opens the KATP channels [15]. The ATP inhibition of KATP channels is powerfully relieved by phosphoinositol diphosphate: formed by α-adrenergic or angiotensin II signals [16]. Long chain fatty acids (LCFA) and their acyl-Co A esters increase the open probability of KATP channels [17]. Because of a higher (> 2 times) single channel conductance (80 pS) and a higher (> 10 times) channel density of KATP channels than those of inwardly rectifying K+ channels (9-45 pS) and delayed rectifier K+ channels (3-10 pS): the activation of only a small fraction of KATP channels can contribute to substantial K+-efflux [18].
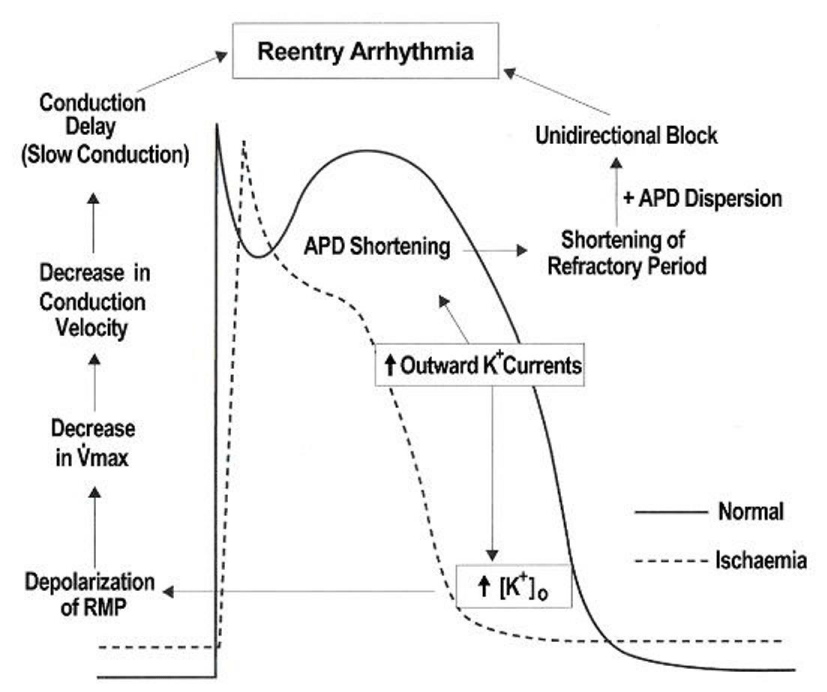
Figure 5 : Mechanisms of reentry arrhythmias.
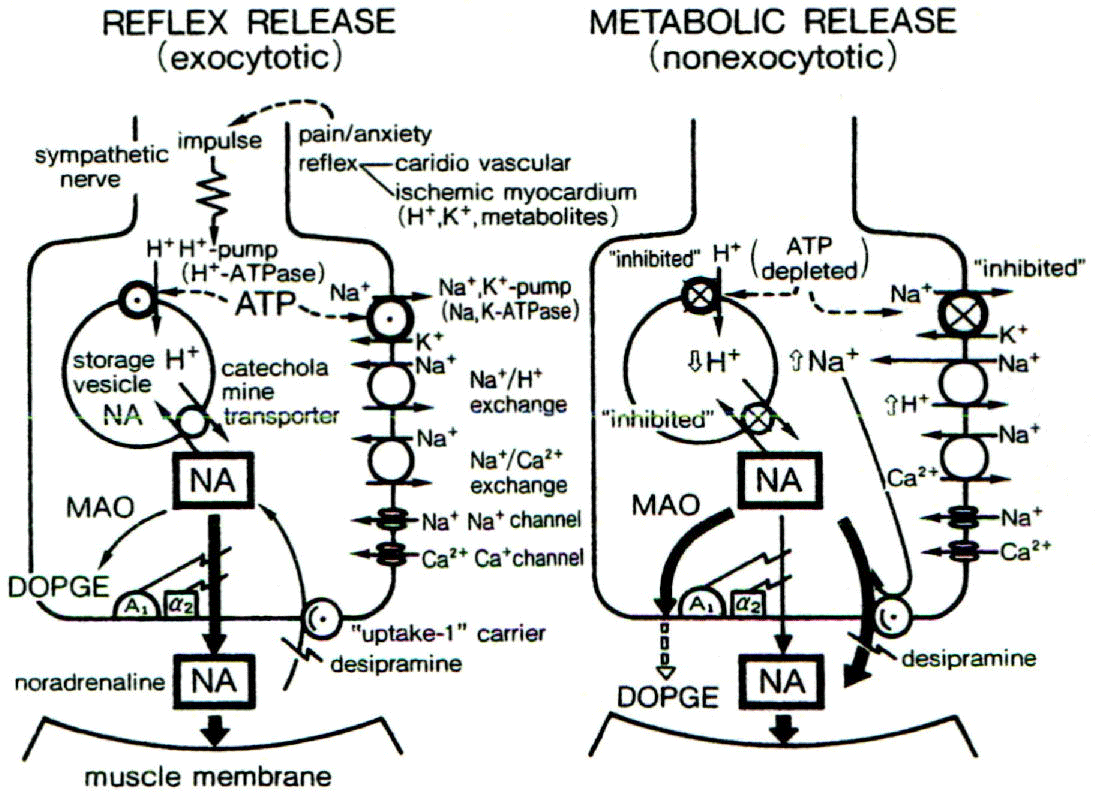
Figure 6 : Noradrenaline release from sympathetic nerve terminals. Courtesy of Dr. M. Arita, Oita, Japan.
Other K+ currents contributing to the increase in [K+]o
Pathological channels
Na+-activated K+ channel current (IK: Na): This channel (~ 200 pS) is activated when intracellular Na+ concentration ([Na+]i) increases from normal (5-10 mM) to > 20 mM. The [Na+]i increases via Na+/H+ exchange in phase I: and via INa/K inhibition in phase III [19]. Fatty acid-activated K+ channel current (IK: FFA): This channel (94 pS) is activated by fatty acid metabolites and amphiphiles that build up during sustained ischemia (phase III) [20].
Modification of physiological channels and exchangers
Ca2+-dependent K+ channel current (IK: Ca): These channels are part of the delayed rectifier K+ super-family: have a very large conductance: and are activated by an increase in intracellular Ca2+ concentration ([Ca2+]i) or stretch [18].
Inwardly rectifying K+ current (IK1): The conductance of these channels increases in very early ischemia (phase I). Up regulation of IK1: with an enhanced inward-rectification: appears within 2~5 min from the onset of hypoxia or metabolic inhibition [21,22]. Intracellular free polyamines: the major factor in the inward rectification: increase in very early ischemia [22]. However: note that: in further ischemia with an increase in [H+]i: the conductance of these channels decreases [13]. Local fall in temperature of more than 1°C during regional ischemia: There is a difference in Q10 values for the amplitude and kinetic parameters of ICa: IKs: IK1 and Ib [23]. This difference may contribute to the decrease in net outward current in the ischemic region. Electrogenic Na+/K+ pump current (INa/K): K+ pumped into the cytoplasm is attenuated during phase III [18]. Na+/Ca2+ exchange current (INa/Ca): Reverse mode of the exchanger carries Na+ out and this outward current shortens the APD [18]. Closing of gap junction channels: i.e. connexins (Cx): In a later stage (> 15 min in phase III): these channels close following a rise in [H+]i: [Ca2+]i or LCFA: and a fall in [ATP]i. This can contribute to slowing of conduction [24,25].
Underlying mechanisms of reentry arrhythmias
A sine qua non for reentry
Malignant ventricular arrhythmias (VF/VT) during early ischemia (phases 1a: 1b) have been interpreted as being due to “reentry arrhythmia”: for which electrical defibrillation is exclusively effective to treat.
An increase in outward K+ currents during early ischemia produces action potential duration (APD) shortening: which inevitably relates to shortening of the effective refractory period (ERP). In addition: APD dispersion occurs between the ischemic: border zone and normal regions. The dispersion of APD and ERP causes unidirectional block. A decrease in resting membrane potential (RMP) suppresses Na+ current (INa): because more Na+ channels are inactivated as the RMP becomes more depolarized: that is: more steady-state inactivation and less availability of the Na+ channels [18]. The depressed INa activated from a depolarized RMP is called “residual Na+ current”: which causes slow conduction. These two conditions and the presence of a reentry pathway are indispensable to the reentry mechanisms [26].
In general: reentry can arise in three pathways: that is: a closed circle: a leading circle and spiral waves. Each of which produces ordered: random and random reentry: respectively. It is supposed that VT is generated from ordered reentry and VF from random reentry [26].
Noradrenalin release from sympathetic nerve terminals during early ischemia
Within the first 5 min of ischemia: noradrenaline (NA) is released (~ 10 times the baseline level) from sympathetic nerve terminals via exocytosis in response to severe pain: anxiety and/or unstable blood pressure. During further ischemia continuing for 10-15 min: [ATP] i decreases in ischemic nerve terminals: which inhibits Na+/K+ pump activity and increases [Na+]i [18,27]. A decrease in the efficiency of proton pumps located on NA storage vesicles further increases the level of NA. In these conditions: the NA uptake-1 carrier functions in reverse mode: which increases presynaptic NA 100-1:000 times the baseline level. This: in turn: produces a second local and “metabolic” (nonexocytotic) NA release [18,27]. The effect of this excessive release of NA is amplified by increases in densities of α- and β-receptor during acute ischemia [18].
Residual Na+ current contributes to the slow conduction
In Figure 7: intracellular cyclic adenosine mono phosphate (cAMP) shifts the h-infinity curve of INa in the negative direction and facilitates the steady-state inactivation of INa [28]. Catecholamines such as NA suppress INa by β1-receptor via cAMP-dependent protein kinase (A-kinase). Thus: in addition to the depolarized RMP: the availability of INa is dramatically decreased during early ischemia: for example: ~80% at -85 mV to ~20% at -75 mV of the RMP. This means residual INa causes “slow conduction” [28].
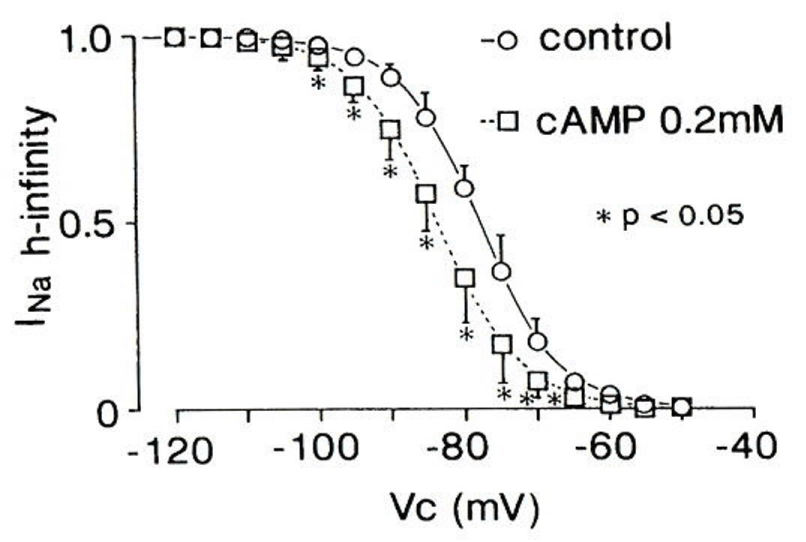
Figure 7 : Intracellular cAMP application decreases INa availability (h-infinity). Modified from [28], with permission of Springer.
Mechanism of VF revealed by means of high-resolution optical mapping
In Figure 8: high-resolution optical mapping of electrical activity with a potentio metric (voltage-sensitive) dye reveals a single: rapidly moving two-dimensional (2D) rotor: that is: a spiral wave. Red denotes the earliest and purple the latest time in the activation sequence (upper two panels). Simultaneous recordings of a surface electrocardiogram (ECG) show turbulent electrical activity. Rapidly moving spiral cores result in irregular ECGs that are characteristic of chaotic VF (bottom two panels) [29].
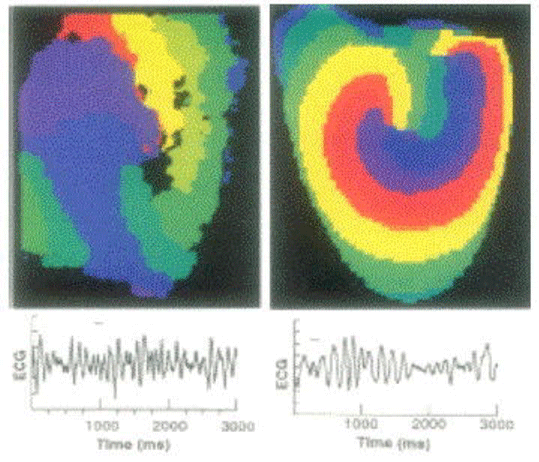
Figure 8 : Isolated rabbit heart experiments and computed isochronal maps. Modified from [29], with permission of the American Association for the Advancement of Science.
In Figure 9: high-resolution mapping of electrical activity reveals the existence of vortex-like activity with a central core that remains excitable. The left panel shows the shape of an Archimedean “spiral wave”. A depolarizing wave front fuses with the re polarizing tail. The spiral wave may produce regular VT. In contrast: the right panel demonstrates “wave breakup”: that is: a change from a spiral wave to “multiple wavelets” [26].
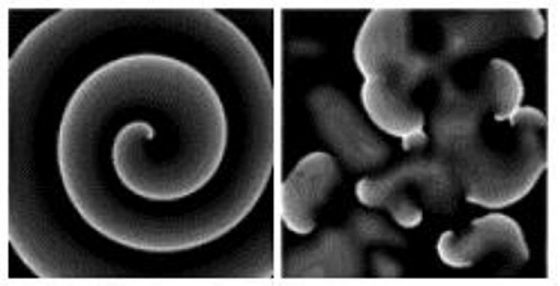
Figure 9 : Spiral wave reentry and wave breakup. Modified from [18], with permission of Springer.
A steep restitution curve (APD / diastolic interval > 1) leads to a large dispersion and alternant of the APD: and promotes the multiple wavelets. The wave breakup means self-perpetuating irregular VF [26,30].
Mechanism of Biphasic Waveform Defibrillation
Defibrillation by monophasic or biphasic waveforms
Figure 10 shows defibrillation threshold (DFT) voltage and energy with various waveforms [31]. Two titanium defibrillation patch electrodes were contoured to fit over the right and left ventricles (epicardium) of the dog heart. VF was induced by an alternating current at 60 Hz. Truncated exponential monophasic or biphasic shock was given 10 sec later: and the DFT was determined (mean + SD). The total duration of the defibrillation waveforms was 10 msec. The biphasic waveform: with the second phase duration equal to (5/5 m sec) or shorter than the first phase: had a significantly lower DFT than the other waveforms. The biphasic waveform: with the shorter first phase than the second phase: had a significantly higher DFT than the other waveforms: including monophasic waveforms [31].

Figure 10 : Defibrillation threshold voltage and energy values for monophasic or biphasic waveforms. Modified from [31], with permission of Wolters Kluwer Health.
In Figure 11: an AP was induced by a monophasic shock (the solid line) and by a biphasic shock (the dashed line) [32]. The hyperpolarizing first phase of the biphasic waveform recruits Na+ channels from their inactivated state: enabling the second phase to induce an AP with prolonged refractoriness. The extended refractoriness has the added benefit of blocking incoming VF wave fronts [32].

Figure 11 : Computer-generated ventricular AP, using the Beeler & Reuter model. Modified from [32], with permission of IEEE.
Na+ channel kinetics and the channel states
Figure 12 shows the time course of INa in correlation with the states of activation and inactivation gates. The model assumes the presence of three identical fast activation gates opening upon depolarization: and one slower inactivation gate closes upon depolarization. Upon depolarization: INa activates within 1 m sec and then inactivates within a few msec. Note that INa inactivates during the depolarization [33,34].

Figure 12 : Hodgkin-Huxley model of independent first order activation and inactivation of Na+ channel current. Modified from [18], with permission of Springer.
Marko processes can explain the state transitions of the Na+ channel (Figure 13). At the RMP of -85 mV: the state of a Na+ channel is either resting (R) or closed (C). Once the membrane is depolarized: the Na+ channel quickly opens (O) and then inactivates (I). When the membrane is re polarized: the inactivated Na+ channel returns to the resting (R) state to prepare for reactivation (O) [34].

Figure 13 : Na+ channel kinetics (state transitions), described by Markow processes.
How a biphasic waveform alters the Na+ channel state
Applying the concept of field stimulation to external defibrillation from the chest wall: a depolarizing (extracellular) pulse hyperpolarizes the (intracellular) cardiac membrane potential: whereas a hyperpolarizing pulse depolarizes it; nevertheless: some unequal responses may exist in a cubic mass of the whole ventricle. With the biphasic waveform: the depolarizing phase (intracellular hyper polarization) resets Na+ channels to their resting state (R) from every state (R: C or I) so they might be reactivated by the second pulse (Figure 14). The hyperpolarizing phase (intracellular depolarization) simultaneously and uniformly activates (O) Na+ channels and subsequently inactivates (I) them. The first phase of the biphasic waveform shock can recruit Na+ channels even from the inactivated state (I): enabling the second phase to induce an AP with prolonged refractoriness [32]. The uniform change in all Na+ channels in either state is critical and effective to defibrillate chaotic VF with random reentry. In order to have a successful defibrillation: more than ~75% of all ventricular cells should be depolarized and inactivated [3].

Figure 14 : Na+ channel state transitions induced by field stimulation with biphasic waveforms.
In contract: a monophasic depolarizing waveform (intracellular hyper polarization) may reset Na+ channels: but it cannot inactivate all the Na+ channels at the end of the single pulse: for example: those in a core of an ordered / random reentry circuit of VT/VF [32,35].
Mechanisms of Restoration of Spontaneous Electrical Activity (Pace Making)
Pacemaker activity is generated from spontaneous diastolic depolarization (phase 4 of AP). In general: pacemaker activity occurs in (1) sinoatrial (sinus) node (SAN) cells: (2) atrioventricular node (AVN) cells: and (3) Purkinje fiber cells [18].
Sino atrial node (SAN)
The SAN is “primary pacemaker” of the heart. Phase 4 is between -60 and -40 mV: depending on the location within the SAN: for example the center (Figure 15) or periphery. The SAN possesses marked redundancy in pacemaker mechanisms. The exact role of each channel is still a matter of debate. However: it is reasonable that many currents contribute to the security of this critical pacemaker function. Several voltage- and time-dependent currents contribute to the phase 4 depolarization. The following are listed in order from more to less negative activation potentials [18,36,37].

Figure 15 : Sinoatrial node (SAN) diastolic depolarization and AP. Modified from [18], with permission of Springer.
- Hyperpolarization-activated (funny) current (Ih(f)).
- Delayed rectifier outward K+ current: rapid or slow component (IKr: IKs).
- Sustained inward current (Ist).
- Background current (Ib).
- T-type Ca2+ current (ICaT).
- L-type Ca2+ current (ICaL).
- Na+/Ca2+ exchange current (INa/Ca).
- TTX-sensitive Na+ current (INa).
Atrioventricular node (AVN)
Conduction through the AVN is slow because APs in the compact portion of the AVN are dependent on the much slower Ca2+ currents [38]. The AVN also can function as a subsidiary pacemaker. Junctional escape rhythms are usually 40-60 beats per a minute (bpm): depending on the pace making site. Anatomically: the AVN is composed of atrionodal (AN): nodal (N): and nodohisian (NH) regions (Figure 16). The AN and NH is the regions bordering the atria and His bundle: respectively. Phase 4 in the N region is -60 to -40 mV and -70 to -60 mV in the AN and NH regions. The N region has essentially the same currents and their contribution to pace making as those in the SAN. In the AN and NH regions: INa contributes to both pace making and the upstroke of the AP (phase 0) [39].

Figure 16 : Atrioventricular node (AVN) diastolic de polarizations and APs. Modified from [26], with permission of Futura Publishing Company.
ICaL: IK and Ist are dependent on ATP and: therefore: sensitive to ischemia. ICaL: IK: Ist: Ih(f) and INa/Ca are increased by β-adrenergic stimulation [18]. These properties are crucial to a return of spontaneous electrical activity during recovery from global ischemia.
Purkinje fiber cells
His bundle and Purkinje fibers have fast conduction velocities. They also function as subsidiary pacemakers. Phase 4 of Purkinje fiber cells is -90 to -60 mV (Figure 17). The idioventricular rhythm of Purkinje fibers is ~30 bpm. Ih is the primary current responsible for phase 4 depolarization. INa contributes to phase 0 as well as the later part of phase 4. β-adrenergic stimulation by adrenaline enhances Ih and produces a positive chronotropic effect [40].

Figure 17 : Purkinje fiber cell diastolic depolarization and AP. Modified from [18], with permission of Springer.
Purkinje fibers are anatomically resistant to ischemia due to their inherent covering sheath. They are also functionally less sensitive to ischemia due to an ample supply of glycogen and fewer myofibrils. Thus: an idioventricular escape rhythm may be the most common pacemaker rhythm after defibrillation: when prolonged ischemia persists. In any case: there is little clinical research on which electrical activity dominates after defibrillating how long VF/VT: and how it improves during resuscitation. Future detailed clinical investigations are warranted.
Global ischemia is a time-dependent process
“Global ischemia” due to untreated VF/VT arrest
In a swine experimental model: VF was induced by rapid pacing with alternating current to the right ventricle (Table 1) [41]. Although the reduction of PO2 was statistically significant: that change was not remarkable after: at least: 5 minutes of the induced VF arrest. Hence: the average estimated SaO2 in arterial blood did not so remarkably desaturate in the early global ischemia [41].
![]()
Before Arrest(mmHg)
After 5 min of Untreated VF (mm Hg)
Arterial
pH
7.44�0.05
7.44�0.13
HCO3
27�3
26�3
PCO2
39�3
40�9
PO2
81�14
69�15*
(estimated SaO2)
(~94.5%)
(~92.5%)
Mixed Venous
pH
7.41�0.05
7.35�0.08*
HCO3
28�3
26�3
PCO2
43�3
48�8*
PO2
40�5
38�8*
*P<0.05, (n=52 ineach group). Modifiedfrom [41],withpermission of Elsevier.
Table 1: Arterial and mixed venous blood gases after 5 minutes of VF arrest without chest compression and ventilation.
Natural time course of VF arrest
In this Holter’s ECG recording: PVC (R on T type) triggers VF (Figure 18). The course VF: with a higher frequency during the first couple of minutes: gradually turns to fine VF with a lower frequency and finally to asystole within ~10 min [3].

Figure 18 : An incidental Holter’s ECG recording of VF to asystole. (UN published data).
Three phase: time-sensitive model/hypothesis of VF/VT arrest
The “Electrical” phase: within approximately 4 min. Early global ischemia is due to almost no coronary flow during the VF/ VT cardiac arrest. [K+]o already starts to increase: which makes additional substrates for reentry mechanisms: such as slow conduction and shortening of ERP: outside the regional ischemia with occluded coronary artery: besides the preexisting more severe ischemic core region. Oxygen saturation in systemic arterial blood is not so remarkably reduced in this early period. The success rate of immediate defibrillation is so high that it restores organized electrical (pace making) activity and sufficient cardiac output (ventricular contraction): that is: a return of spontaneous circulation (ROSC). Thus: a transient (≤~4 min) global ischemia is reversible [42]. The “Hemodynamic” phase: from approximately 4 to 10 min. Anaerobic glycolytic ATP synthesis may contribute to maintaining sub sarcolemmal ATP levels: though intracellular acidosis proceeds. To stop the global ischemia: oxygen must be delivered; therefore: chest compression and appropriate ventilation are critical to defibrillation. The removal of accumulated deleterious ischemic metabolic factors is also important [42]. The “Metabolic” phase: after approximately 10 min.
[ATP]issue injury/stunning (necrosis/apoptosis) from the prolonged global ischemia and reperfusion is accompanied by a decrease in [ATP]i: increases in [Ca2+]i and [Na+]i: and the appearance of an oxidant burst and polyunsaturated lipids peroxide [42]. Experiments designed to test the beneficial effects of mild therapeutic hypothermia: and future investigations of chemical and immunological mechanisms are warranted.
Reperfusion Injury after Global Ischemia
Stunned myocardium
Reperfusion injury occurs after 10 min of global ischemia in a rat heart (Figure 19) [43]. The reperfusion injury contains the following phenomena. Mechanical stunning: i.e. contractile disturbance of the ventricle. Reperfusion arrhythmias (no sustained VT or VT/ VF). Approximately 75% of no sustained VT (NSVT) is caused by non-reentry mechanisms: that is: triggered activity or delayed after depolarization’s (DAD). Micro vascular damage: that is: excessive formation of peroxynitrite from nitric oxide (. NO). Stunning is reversible if reperfusion occurs after 5-20 min of regional or global ischemia. Contractile failure with reduced developed tension and increased resting tension reflect the “stunned myocardium”: which can contribute to post-defibrillation pseudo pulse less electrical activity (PEA) if the previous VF/VT arrest induces sufficient global ischemia. Hypothetically: [Ca2+]i overload and oscillations damage the contractile proteins too much to normally respond to Ca2+ [38,43]. Stunned myocardium can respond well to inotropic stimulation by catecholamines. For example: adrenaline (epinephrine) is the first-line drug used for PEA: according to the ACLS algorithm [1,43].

Figure 19 : Postischemic early reperfusion injury. Modified from [43], with permission of The American Physiological Society.
KATP channels are activated not only during ischemia: but also during an early phase after reperfusion. The opening of KATP channels protects the stunned myocardium (“memory effect”): probably by shortening the APD and decreasing Ca2+ overload in the myocardium [44]. The shorter the duration of global ischemia: the milder the ischemic damage and the less severe the reperfusion injury and contractile disturbance.
Increase in [Ca2+]i in stunning after early reperfusion
Cytosolic Ca2+ overload is likely to involve the following (Figure 20). Na+/H+ exchange and Na+/Ca2+ exchange: Rapid washout of extracellular H+ activates the Na+/H+ exchanger: bringing more Na+ into the cells. The reverse mode of Na+/Ca2+ exchange carries Na+ out and Ca2+ in while Na+/K+ pump activity is suppressed [45]. Entry through Ca2+ channels: Ca2+ channel blockers are effective in attenuating the stunning and Reperfusion arrhythmias [38]. Decreased Ca2+ uptake into the sarcoplasmic reticulum (SR): Reduced availability of ATP also decreases Ca2+ extraction from the cells [38]. Burst of oxygen-derived free radicals and oxidative stress: The formation of reactive oxygen species (ROS) promotes Ca2+ release from the SR. The hydroxyl radical (. OH): the most aggressive ROS: peroxidates polyunsaturated lipids: which increases membrane permeability to Ca2+ [38]?

Figure 20 : Mechanisms of Ca2+ overload, Ca2+ modulation, and free radicals. Courtesy of Dr. M. Yasutake, Tokyo, Japan.
Major sources of the ROS include the following [38].
- Complex III (IV: I) in the electron transport chain of mitochondria (physiological sources).
- Nicotinamide adenine dinucleotide phosphate (NADPH: reduced form) oxidase system.
- Nitric oxide (NO) from the endothelium: when in excess: forms peroxynitrite (. NO).
Conclusion
Within the first 10 min of AMI: K+ conductance and [K+] o increase: without remarkable depletion of bulk [ATP]i (> 2 mM). VF/VT arrest: associated with ACS: commonly occurs during this period due to arrhythmogenic substrates. Additionally: arterial blood oxygen does not desaturate remarkably during the “electrical” phase of VF/VT arrest. Depolarization of the myocardial RMP and shortening and spatial dispersion of APD are the basis for reentry: the main mechanism of VF/VT during early ischemia. Such reentry occurs under conditions of unidirectional block and slow conduction due to the reduced (residual) INa.
Arrhythmogenic substrates of ischemia mainly involve hypoxia/ anoxia: glucose depletion: intracellular acidosis and extracellular hypokalemia. Associated substrates include increased intracellular Na+ and Ca2+: noradrenalin secretion from nerve terminals: an increase in amphipathic substances: intrinsic inflammatory factors: and local hypothermia. Ischemia-induced changes in ion channel currents are directly responsible for the reentry. Although IK: ATP is pivotal: IK: Ca: IK: Na: IK: FFA and INSC also play a role. Increase in K+ conductance and the outward K+ currents shorten APD and depolarize RMP with an increase in [K+]o. An inhomogeneous distribution of channel currents and their modification during ischemia facilitate APD dispersion. Inhibition of INa/K: and the reverse mode of INa/Ca is also involved in the initiation of reentry. Decreased availability of INa (residual INa): due to a depolarized RMP: and inhibition of gap junction (Cx) channel current cause slowed conduction: another essential factor of reentry.
High-resolution optical mapping of electrical activity using voltage-sensitive dyes reveals the VT reentry as vortex-like activity with a central core. Breakup of a spiral wave (regular VT) into multiple wavelets causes self-perpetuating chaotic VF (random reentry).
Biphasic waveforms are more effective than monophonic waveforms to defibrillate VF during ischemia. The first (hyperpolarizing) pulse resets Na+ channels to the resting state: allowing them to be reactivated by the second (depolarizing) pulse. This depolarizing pulse simultaneously and uniformly depolarizes almost all the Na+ channels and enables them to induce an AP with prolonged refractoriness. The uniform change in almost all channels in either state is crucial and effective to defibrillate VF with random reentry. In contrast: a monophasic depolarizing waveform cannot reactivate the inactivated Na+ channels in the core of an ordered/ random reentry circuit of VF/VT.
After electrical defibrillation of the VF/VT: pacemaker activity may be reclaimed from the SAN or: alternatively: by escape rhythms from the AVN or Purkinje cells: depending on the severity of the ischemic conditions. Spontaneous diastolic depolarization (phase 4) produces the pacemaker activity of cells in the conduction pathway: with the pacing rate highest in the SAN. Pacemaker-related currents during phase 4 are complicated and still a matter of debate. Pacemaker cells in the SAN possess both rapid and slow components of IK (IKr: IKs): ICaT: ICaL: Ih(f): Ib: Ist: and electrogenic INa/K. The latter part of the phase 4 depolarization and transition to the AP upstroke (phase 0) are affected by ICaT: inward INa/Ca and TTX-sensitive INa.
Although the AVN and Purkinje cells have essentially the same currents as those in SAN cells: the contributions of ICaL and Ih(f) are progressively larger in the AVN and Purkinje fibers because of their more negative maximum diastolic potentials. Among the pacemaker currents: IK: ICaL: Ist and Ih(f) strongly depend on metabolic conditions and are very sensitive to ischemia. Therefore: pacemaker activity during ischemia may be more suppressed in the SAN and AVN than in Purkinje cells: where those currents are larger under normal conditions.
Post ischemic reperfusion injury: that is: stunned myocardium and contractile failure: can occur after even short-term regional and/or global ischemia. The stunning due to increased [Ca2+]i can result in post-defibrillation pseudo PEA: and hinder or even upset a ROSC if spontaneous electrical activity is restored after the successful defibrillation. [Ca2+]i overload and oscillations damage the contractile proteins too much to respond normally to Ca2+. The Ca2+ overload is mainly due to enhanced Na+/H+ exchange resulting in reverse mode Na+/Ca2+ exchange: a burst of oxygen-derived free radicals: and oxidative stress.
The mechanisms of VF/VT arrest during ischemia and the restoration of pacemaker activity after defibrillation are reviewed briefly with an emphasis on ion channels: transporters and molecular mechanisms. Continuous high-quality chest compression early after VF/VT arrest and immediately after defibrillation maintains coronary as well as cerebral and systemic blood perfusions. Such resuscitation suppresses global ischemia in the whole heart: decreases arrhythmogenic substrates: washes out the ischemic related products: and provides oxygen and glucose for pacemaker and ventricular cells to pace and contract. Thus: an understanding of the cellular pathophysiology that underscores the need for continuous high-quality chest compression is essential for BLS/ACLS education: training and clinical practice.
Acknowledgement
An author (H.M.) gratefully acknowledges the generosity of Professor Makoto Arita and Professor Richard D. Nathan in supporting cardiac electrophysiological research and editing the manuscript: and the encouragement of Professor Teruo Takano in BLS/ACLS education.
For permission to reproduce copyrighted figures and a table: we would like to thank the following publishers and societies. American Association for the Advancement of Science (Science): Figures 2 and 8; Elsevier (Resuscitation): Table 1; Elsevier (J Mol Cell Cardiol): Figure 1; Futura Publishing Company (Basic Cardiac Electrophysiology for the Clinician): Figure 16; IEEE (IEEE Trans Biomed Eng): Figure 11; Oxford University Press (Cardiovasc Res): Figure 3; Springer (Pfl�gers Arch): Figure 7; Springer (Cardiac Cellular Electrophysiology): Figure 9: Figure 12: Figure 15: Figure 17; The American Physiological Society (Am J Physiol): Figure 19; Wolters Kluwer Health (Circ Res): Figure 4; Wolters Kluwer Health (Circulation): Figure 10.
References
- Field JM. The Textbook of Emergency Cardiovascular Care and CPR. Philadelphia: Lippincott Williams & Wilkins. 2009.
- Cummins RO, Field JM, Hazinski MF. ACLS: Principals and Practice – The Reference Textbook. Dallas: American Heart Association. 2003.
- Norman AP, Henry RH, Karl BK, Volker W, Douglas AC. Cardiac Arrest: the Science and Practice of Resuscitation Medicine. 2nd edn. Cambridge: Cambridge University Press. 2007.
- Kléber AG. Extracellular potassium accumulation in acute myocardial ischemia. J Mol Cell Cardiol. 1984; 16: 389-394.
- Harris AS, Bisteni A, Russell RA, Brigham JC, Firestone JE. Excitatory factors in ventricular tachycardia resulting from myocardial ischemia; potassium a major excitant. Science. 1954; 119: 200-203.
- Smith WT, Fleet WF, Johnson TA, Engle CL, Cascio WE. The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Experimental Cardiology Group, University of North Carolina. Circulation. 1995; 92: 3051-3060.
- Wilde AA, Aksnes G. Myocardial potassium loss and cell depolarisation in ischaemia and hypoxia. Cardiovasc Res. 1995; 29: 1-15.
- Wilde AA, Escande D, Schumacher CA, Thuringer D, Mestre M, Fiolet JW, et al. Potassium accumulation in the globally ischemic mammalian heart. A role for the ATP-sensitive potassium channel. Circ Res. 1990; 67: 835-843.
- Allen DG, Morris PG, Orchard CH, Pirolo JS. A nuclear magnetic resonance study of metabolism in the ferret heart during hypoxia and inhibition of glycolysis. J Physiol. 1985; 361: 185-204.
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983; 305: 147-148.
- Priebe L, Friedrich M, Benndorf K. Functional interaction between K(ATP) channels and the Na(+)-K(+) pump in metabolically inhibited heart cells of the guinea-pig. J Physiol. 1996; 492 : 405-417.
- Schackow TE, Ten Eick RE. Enhancement of ATP-sensitive potassium current in cat ventricular myocytes by beta-adrenoreceptor stimulation. J Physiol. 1994; 474: 131-145.
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999; 79: 917-1017.
- Katz AM. Physiology of the Heart, 4th edn. Philadelphia: Lippincott Williams & Wilkins. 2004.
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol. 1999; 61: 337-362.
- Okamura M, Kakei M, Ichinari K, Miyamura A, Oketani N, Koriyama N, et al. State-dependent modification of ATP-sensitive K+ channels by phosphatidylinositol 4,5-bisphosphate. Am J Physiol Cell Physiol. 2001; 280: C303-308.
- Liu GX, Hanley PJ, Ray J, Daut J. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to K(ATP) channels in the heart. Circ Res. 2001; 88: 918-924.
- Carmeliet E, Vereecke J. Cardiac Cellular Electrophysiology. Norwell: Kluwer Academic Publishers. 2002.
- Kameyama M, Kakei M, Sato R, Shibasaki T, Matsuda H, Irisawa H. Intracellular Na+ activates a K+ channel in mammalian cardiac cells. Nature. 1984; 309: 354-356.
- Kim D, Clapham DE. Potassium channels in cardiac cells activated by arachidonic acid and phospholipids. Science. 1989; 244: 1174-1176.
- Muramatsu H, Sato R, Okumura H. Early increase in K+ conductance during metabolic inhibition by cyanide in guinea pig ventricular myocytes. Nihon Ika Daigaku Zasshi. 1990; 57: 308-321.
- Piao L, Li J, McLerie M, Lopatin AN. Cardiac IK1 underlies early action potential shortening during hypoxia in the mouse heart. J Mol Cell Cardiol. 2007; 43: 27-38.
- Kiyosue T, Arita M, Muramatsu H, Spindler AJ, Noble D. Ionic mechanisms of action potential prolongation at low temperature in guinea-pig ventricular myocytes. J Physiol. 1993; 468: 85-106.
- Noma A, Tsuboi N. Dependence of junctional conductance on proton, calcium and magnesium ions in cardiac paired cells of guinea-pig. J Physiol. 1987; 382: 193-211.
- Yamada KA, McHowat J, Yan GX, Donahue K, Peirick J, Kl�ber AG, et al. Cellular uncoupling induced by accumulation of long-chain acylcarnitine during ischemia. Circ Res. 1994; 74: 83-95.
- Jalife J. Basic Cardiac Electrophysiology for the Clinician. Armonk: Futura Publishing Company. 1999.
- Kurz T, Richardt G, Hagl S, Seyfarth M, Sch�mig A. Two different mechanisms of noradrenaline release during normoxia and simulated ischemia in human cardiac tissue. J Mol Cell Cardiol. 1995; 27: 1161-1172.
- Muramatsu H, Kiyosue T, Arita M, Ishikawa T, Hidaka H. Modification of cardiac sodium current by intracellular application of cAMP. Pflugers Arch. 1994; 426: 146-154.
- Gray RA, Jalife J, Panfilov AV, Baxter WT, Cabo C, Davidenko JM, et al. Mechanisms of cardiac fibrillation. Science. 1995; 270: 1222-1223.
- Weiss JN, Chen PS, Qu Z, Karagueuzian HS, Garfinkel A. Ventricular fibrillation: how do we stop the waves from breaking? Circ Res. 2000; 87: 1103-1107.
- Dixon EG, Tang AS, Wolf PD, Meador JT, Fine MJ, Calfee RV, et al. Improved defibrillation thresholds with large contoured epicardial electrodes and biphasic waveforms. Circulation. 1987; 76: 1176-1184.
- Jones JL, Jones RE, Milne KB. Refractory period prolongation by biphasic defibrillator wave-forms is associated with enhanced sodium current in a computer model of the ventricular action potential. IEEE Trans Biomed Eng. 1994; 41: 60-68.
- HODGKIN AL, HUXLEY AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952; 116: 449-472.
- Hille B. Ionic Channels of Excitable Membranes. 2nd edn. Massachusetts: Sinauer Associates.1992.
- Jones JL, Tovar OH. Electrophysiology of ventricular fibrillation and defibrillation. Crit Care Med. 2000; 28: N219-221.
- Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993; 73: 197-227.
- Muramatsu H, Zou AR, Berkowitz GA, Nathan RD. Characterization of a TTX-sensitive Na+ current in pacemaker cells isolated from rabbit sinoatrial node. Am J Physiol. 1996; 270: H2108-2119.
- Opie LH. Heart Physiology: From Cell to Circulation. 4th edn. Philadelphia: Lippincott Williams & Wilkins. 2004.
- Munk AA, Adjemian RA, Zhao J, Ogbaghebriel A, Shrier A. Electrophysiological properties of morphologically distinct cells isolated from the rabbit atrioventricular node. J Physiol. 1996; 493 : 801-818.
- Di Francesco D. A new interpretation of the pace-maker current in calf Purkinje fibres. J Physiol. 1981; 314: 359-376.
- Tucker KJ, Idris AH, Wenzel V, Orban DL. Changes in arterial and mixed venous blood gases during untreated ventricular fibrillation and cardiopulmonary resuscitation. Resuscitation. 1994; 28: 137-141.
- Weisfeldt ML, Becker LB. Resuscitation after cardiac arrest: a 3-phase time-sensitive model. JAMA. 2002; 288: 3035-3038.
- Meissner A, Morgan JP. Contractile dysfunction and abnormal Ca2+ modulation during postischemic reperfusion in rat heart. Am J Physiol. 1995; 268: H100-111.
- Shigematsu S, Sato T, Abe T, Saikawa T, Sakata T, Arita M. Pharmacological evidence for the persistent activation of ATP-sensitive K+ channels in early phase of reperfusion and its protective role against myocardial stunning. Circulation. 1995; 92: 2266-2275.
- Yasutake M, Ibuki C, Hearse DJ, Avkiran M. Na+/H+ exchange and reperfusion arrhythmias: protection by intracoronary infusion of a novel inhibitor. Am J Physiol. 1994; 267: H2430-2440.