
Case Report
Austin J Cerebrovasc Dis & Stroke. 2014;1(5): 1021.
Adult Phenotypic Spectrum of Headache, Myopathy and Ischemic Stroke Associated with Mitochondrial POLG Mutation
Jiangyong Min*, Muhammad Umar Farooq and Christopher Glisson
Mercy Health Hauenstein Neuroscience, Grand Rapids and Michigan State University, Michigan
*Corresponding author: Jiangyong Min, Hauenstein Neuroscience Center, Mercy Health St Mary’s Hospital, Grand Rapids, Michigan, 220 Cherry Street SE, Grand Rapids, 49503, Michigan
Received: August 20, 2014; Accepted: September 17, 2014; Published: September 19, 2014
Abstract
Mutations in the POLG gene have emerged as one of the most common causes of inherited mitochondrial disease. We reported a rare case presenting with adult phenotypic spectrum of headache, myopathy, and ischemic stroke associated with POLG1 heterozygote mutation. This should be classified as a distinct clinical entity rather than an atypical MELAS syndrome.
Case Presentation
A 59 years old Caucasian female was admitted to the neurology service in July, 2013 for new onset left sided weakness and unspecified paresthesia associated with visual disturbance. Neurological exam was essentially unremarkable except for subtle left arm weakness without pronator drift. A brain MRI scans revealed sub acute ischemic infarction involving the left occipital and parietal lobes (Figure 1). Conventional angiogram revealed, “An incident find of approximately 2.5mm focal region of out pouching at the right posterior communicating artery origin; determined to most likely be an infundibulum. Posterior communicating artery appears to arise from the apex of this out punching. No significant hemodynamic stenosis intracranially or extracranially was found”. A repeated CT angiogram taken seven days later presented consistent results as before, “no significant cervical or intracranial artery abnormality. No significant change appreciated with regard to posterior communicating artery infundibulum seen on recent conventional angiogram”. Transesophageal echocargiogram showed normal cardiac function with no evidences for cardiac thrombus, patent foramen ovale, or atrial septal defect. Hypercoagulable state workup and 28-day prolonged cardiac monitoring were negative. She had sixteen pregnancies and five live births. In addition to carrying a history of controlled hypertension and uncontrolled hyperlipidemia (total cholesterol 228 mg/dl, triglycerides 201 mg/dl, high density lipid 44 mg/dl and low density lipid 144 mg/dl), the patient reported a plethora of previous medical courses.
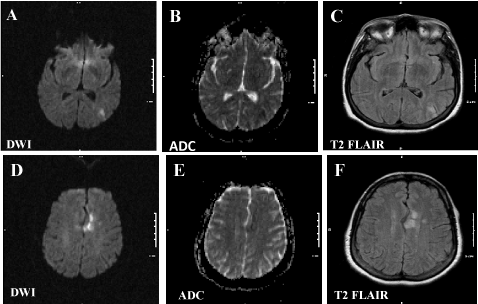
Figure 1 : A brain MRI scan revealed sub acute infarctions involving the left occipital (A-C) and parietal lobes (D-F) as hyper intensity signals in restricted diffused imaging (DWI) with corresponding hypo intensity signals in apparent diffusion coefficient (ADC). The ischemic lesions are not confined to vascular territories. T2 FLAIR: T2 fluid-attenuated inversion recovery.
Over the past ten years, she developed progressive broad spectrum nonspecific neurological symptoms including: easy to fatigue, intermittent paresthesia in the limbs, headaches with migraine features, visual disturbances “seeing spots and lights, tinnitus, episodic confusion and dysarthria”, and mild cognitive and memory impairment. She had a muscle biopsy in 2006 due to muscle weakness with unknown etiology, which reported a ragged red fiber and 5cytochrome c Oxidase (COX) negative fibers. The common mtDNA mutations were screened; all reported negative results. There has been a myasthenia gravis (MG) evaluation that was also negative including negative anti-MUSK antibody. In July 2007 she had extensive electro diagnostic examinations of the left lower and upper extremities, facial muscle, and paraspinal muscles with no evidence of a generalized myopathy; incomplete motor unit potential activation in nearly all muscles studied that may be associated with incomplete voluntary efforts, a central disorder of motor unit control, or pain; nonspecific abnormalities of motor unit potential recruitment in several facial muscles. These electromyography (EMG) findings are suggestive of a neurogenic firing pattern, but it is non diagnostic and of questionable clinical significance. The nuclear-encoded DNA polymerase γ (POLG) gene testing in 2009 did show a heterozygote mutation; Y831C. This POLG mutation is reported as an autosomal dominant gene that could cause chronic progressive ophthalmolegia and Parkinson’s [1,2]. Several Electro Encephalo Graphy.
(EEG) tests for episodic confusion showed normal awake and asleep results. Complete Blood Count (CBC) and biochemistry (including lactate serum level, organic acids, and amino acids) were all within normal limits. Skeletal muscle mitochondria oxidative phosphorylation test and gene screening for the acid maltase gene in December 2009 reported normal. Although test did find the alpha-1,4 glucosidase activity (Pompe disease) to be slightly below the normal limit but deemed to be nonpathogenic since her Pompe gene showed normal sequencing results. Inflammatory and autoimmune markers were also normal. Serial ophthalmological exams were normal except chronic posterior vitreous detachment Oculus Uterque (OU). Most recent optical coherence tomography in April 2013 was normal without evidence of optic nerve atrophy.
During the months following her stroke in July of 2013, she developed congestive heart failure as well as slightly worsening fatigue. However, her recent neurology visit in January 2014 continues to show a lack of focal neurological deficits. She shows no signs of fatigability and myotonic phenomena; cognitive impairment was not detected per bedside Mini�Mental State Examination (MMSE).
Discussion
The nuclear-encoded DNA polymerase γ (POLG) is the sole DNA polymerase responsible for the replication and repair of the mitochondrial genome [3,4]. Pol γ has both polymerase and 3’-5’ proffreading exonuclease activity, and is translated from 22 exons of the POLG1 gene on chromosome 15q25 [5]. The first pathogenic mutations of POLG1 were identified in families with autosomal dominant chronic Progressive External Ophthalmoiplegia (PEO) associated with the accumulation of multiple mitochondrial DNA 9mtDNA) deletions in clinically affected tissues [6]. Subsequently, reports have shown that mutations in subunit 1 (POLG1) gene are directly linked to defects in mitochondrial RNA (mtRNA) [7,8], leading to Alpers syndrome [9,10], sensory ataxia, neuropathy, dysarthria ophthalmoparesis (SANDO) [11], and epilepsy syndrome [12]. However the specific pathogenic status of various mutations remains unclear due to conflicting reports on their frequencies in populations of healthy individuals. For instance, the Y831C mutation was first reported by Barthelemy [1] while examining a patient experiencing severe mtRNA depletion. However, despite the significant change (from a tyrosine to a cysteine) of an amino-acid in the catalytic subunit of POLG, the authors ruled this mutation to be non-pathogenic because it was located outside the functional region of the protein, involved a poorly conserved amino acid and was present not only in patient but also in healthy subjects such as the patient’s mother and grandmother. In contrast, Mancuso et al. [2] reported a case of the same Y831C mutation to be the cause of an entire family’s PEO, peripheral neuropathy and Parkinsonism. Mancuso and colleagues indicated that the Y831C mutation was not found in 130 healthy subjects in their study. These conflicting reports suggest that mutations similar to Y831C of POLG could be contributing factors to, but not necessarily the sole cause of neurological disorders related to mtRNA. With the crystal structure of human POLG holoenzyme now solved [13], our ability to predict the effects of POLG mutations should improve dramatically in the future.
Deschauer and colleagues [14] reported a case of a patient who presented stroke like episodes, headaches, seizures, and a right occipital lesion. Muscle biopsy demonstrated ragged-red fibers and genetic study revealed heterozygous mutations in the POLG1 gene. This evidence led the authors to conclude that MELAS (mitochondrial encephalopathy, lactic acidosis and stroke-like episodes) could be included in symptoms indicative of POLG1 mutations. POLG1 mutation and MELAS currently have different clinical presentations with many overlapping symptoms. The patient in Deschauer’s report has very similar clinical presentations to our patient discussed here, who also has episodic headaches and encephalopathy as well as stroke like symptoms. Her brain MRI ischemic lesions do not fit the typical vascular distribution. These presentations are similar to the clinical phenotypes of MELAS. However, our patient has atypical aspects as for MELAS. Her symptoms started in her 40s rather than the early onset that is typical of MELAS. CSF study showed no signs of elevated lactic acidosis. MELAS can result from mutations in one of several genes, includingMT-ND1,MT-ND5,MT-TH,MT-TL1, and MT-TV [15]. MT-TL1, cause the majority of MELAS cases. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy that would subsequently lead to a chronic state of insufficient cellular energy although exact mechanisms of how this genetic mutation causes clinical phenotype of MELAS [16-18]. Somewhat recently, another case was reported [19] where a patient’s clinical presentation reflects some of the MELAS phenotypes including migraine headaches, generalized weakness, and asymmetric ischemic stroke involving occipital lobe. However, the patient’s serum and CSF lactic acid were normal, and carried no known phenotypic mutations of MLEAS. All of these are similar to our patient, who tested 5 COX negative but showed positive results for ragged red fiber in a muscle biopsy and POLG1 heterozygote mutation suggesting mitochondrial disease. Most of her mutations are reported to be heterogeneous and only a few of these mutations have been identified in non-related cases, making it extremely difficult to make any definitive mutation to phenotype links.
We disagree with the current established notion that this is a MELAS associated disorder with mutations in the POLG1 gene [14]. As with a similar case reported recently by Cheldi and colleagues [19], we recommend that these cases should be classified as a distinct clinical entity rather than an atypical MELAS syndrome. A suspicion of POLG mutation should be warranted if patients with ischemic stroke have similar presentations of MLEAS but lacking of typical clinical and genetic phenotypes of MELAS.
References
- Barthélémy C, de Baulny HO, Lomb�s A. D-loop mutations in mitochondrial DNA: link with mitochondrial DNA depletion? Hum Genet. 2002; 110: 479-487.
- Mancuso M, Filosto M, Oh SJ, DiMauro S. A novel polymerase gamma mutation in a family with ophthalmoplegia, neuropathy, and Parkinsonism. Arch Neurol. 2004; 61: 1777-1779.
- Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982; 28: 693-705.
- Ropp PA, Copeland WC. Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase gamma. Genomics. 1996; 36: 449-458.
- Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004; 73: 293-320.
- Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001; 28: 211-212.
- DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004; 1658: 80-88.
- Longley MJ, Graziewicz MA, Bienstock RJ, Copeland WC. Consequences of mutations in human DNA polymerase gamma. Gene. 2005; 354: 125-131.
- Davidzon G, Mancuso M, Ferraris S, Quinzii C, Hirano M, Peters HL, et al. POLG mutations and Alpers syndrome. Ann Neurol. 2005; 57: 921-923.
- Naviaux RK, Nguyen KV. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol. 2004; 55: 706-712.
- Goethem GV, Martin JJ, Dermaut B, Lofgren A, Wibail A, Ververken D, et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscular Dis. 2003; 13: 133-142.
- Engelsen BA, Tzoulis C, Karlsen B, Lillebø A, Laegreid LM, Aasly J, et al. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain. 2008; 131: 818-828.
- Lee YS, Kennedy WD, Yin YW. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell. 2009; 139: 312-324.
- Deschauer M, Tennant S, Rokicka A, He L, Kraya T, Turnbull DM, et al. MELAS associated with mutations in the POLG1 gene. Neurology. 2007; 68: 1741-1742.
- DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013; 9: 429-444.
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990; 348: 651-653.
- Flossmann E. Genetics of ischaemic stroke; single gene disorders. Int J Stroke. 2006; 1: 131-139.
- Flossmann E. Genetics of ischaemic stroke; single gene disorders. Int J Stroke. 2006; 1: 131-139.
- Cheldi A, Ronchi D, Bordoni A, Bordo B, Lanfranconi S, Bellotti MG, et al. POLG1 mutations and stroke like episodes: a distinct clinical entity rather than an atypical MELAS syndrome. BMC Neurol. 2013; 13: 8.