
Review Article
Austin J Cerebrovasc Dis & Stroke. 2014;1(6): 1026.
Pre-Clinical Models of Acquired Neonatal Seizures: Differential Effects of Injury on Function of Chloride Co-Transporters
Kang SK1 and Kadam SD1,2*
1Neuroscience Laboratory, Hugo Moser Research Institute, USA
2Department of Neurology, Johns Hopkins University, USA
*Corresponding author: Kadam SD, Department of Neurology, Johns Hopkins University School of Medicine, 716 North Broadway, # 426, Baltimore, MD 21205, USA
Received: August 11, 2014; Accepted: October 07, 2014; Published: October 09, 2014
Abstract
Hypoxic-Ischemic Encephalopathy [HIE] represents the most common acquired pathology associated with neonatal seizures. HIE-associated neonatal seizures are often difficult to control, due to their refractoriness to traditional anti-seizure agents. Developmentally regulated chloride gradients during early development make the neonatal brain more seizure-susceptible by depolarizing GABAAR-mediated currents, and therefore hindering inhibition by conventional anti-seizure drugs such as Phenobarbital [PB] and benzodiazepines. Pharmaco-modulation of chloride co-transporters has become a current field of research in treating refractory neonatal seizures, and the basis of two clinical trials [NCT01434225; NCT00380531]. However, the recent termination of NEMO study [NCT01434225] on bumetanide, an NKCC1 antagonist, suggests that clinical utilization of bumetanide as an adjunct to treat neonatal seizures with PB may not be a viable option. Hence, re-evaluation of bumetanide as an adjunct through pre-clinical studies is warranted. Additionally, the model-specific variability in the efficacy of bumetanide in the pre-clinical models of neonatal seizures highlights the differential consequences of insults used to induce seizures in each pre-clinical model as worth exploration. Injury itself can significantly alter the function of chloride co-transporters, and therefore the efficacy of anti-seizure agents that follow.
Keywords: Neonatal seizures; Hypoxic Ischemic
Introduction
Seizures are detected much more frequently during the neonatal period than at any other age [1,2]. Untreated or poorly-controlled seizures in neonates can lead to adverse neuro developmental morbidity and lethality [3-5], and the causative acquired pathologies include intracranial hemorrhage, intracranial infection and Hypoxic- Ischemic Encephalopathy [HIE] [6,7]. HIE has remained the most common underlying pathology, constituting 50-60% of total seizures reported in neonates [7,8]. HIE-associated seizures in neonates display pharmaco-resistance to conventional and first-line antiseizure drugs such as Phenobarbital [PB]and benzodiazepines, which act as agonists to enhance GABAAR-mediated inhibition [2,9,10]. Therapeutic hypothermia has been reported to benefit treatment protocols of anti-seizure pharmacotherapies in neonates [11,12], and is being widely investigated. However, efficacious alternative pharmacotherapies to treat seizures in neonates and bypass the pharmaco-resistance are lacking [9,13]. An attempt to remedy this lack has been made using the NKCC1 blocker, bumetanide, based on the promising data from several pre-clinical models, and two clinical trials [NCT01434225; NCT00380531]were initiated in 2009 to investigate the anti-seizure efficacy of this drug as an adjuvant therapy towards PB-resistant neonatal seizures associated with HIE [14]. However, recent termination of NEMO clinical trial [NCT01434225] that reported non-efficacy of bumetanide and its ototoxicity suggests a need for re-evaluation of potential therapeutic benefit of bumetanide as an anti-seizure adjunct [15].
Intrinsic neuronal hyperexcitability of the immature brain results in a higher seizure susceptibility
The imbalance of excitation and inhibition in a seizing immature brain has been attributed to many factors such as developmentally regulated protein expression and pathology-induced inflammatory response, which ultimately results in neuronal hyperexcitability that leads to the higher seizure susceptibility in neonates [16-18]. During the critical period of synaptogenesis, the developmental regulation of GluR subunits in neurons and glia lowers the threshold for seizures and excitotoxic hypoxic-ischemic injury [16]. Additionally, alteration of proteins such as gephyrin and GABAAR that are crucial for GABA ergic signaling has been proposed as a possible mechanism that results in a diminished inhibition shown in a model with pilocarpine-induced status epilepticus [20].
Another potential factor that has recently emerged is an altered function of chloride co-transporters in immature brain that leads to a reversed movement of chloride ions [Cl-] [21,22]. The neuronal chemical gradient of Cl- is largely driven by membrane-localized chloride co-transporters, specifically Na+K+2Cl- Co-transporter 1 [NKCC1] [23] and K+Cl- Co-transporter 2 [KCC2] [24,25]. NKCC1, present in multiple cell types [26], pumps in Cl-and leads to its intracellular accumulation, whileKCC2, known to be expressed specifically in the CNS [27], extrudes Cl- to maintain a lowintracellular Cl- concentration, which allows for the inhibitory action of GABAAR agonists. In the mature brain, dominant expression of KCC2 over NKCC1 establishes a Cl-gradient that drives Cl-influx upon GABAAR-mediated activation of chloride channels, rendering GABA hyperpolarizing. In contrast, in immature neurons where KCC2 expression is lacking, insufficient Cl- extrusion leads to a high Cl-concentration, resulting in less inhibition or even depolarization in response to GABA. Therefore, pharmaco-modulation of chloride co-transporters to lower Cl- concentrations in immature brains to allow GABAAR-mediated inhibition has been investigated as a promising therapeutic intervention [28,29].
Bumetanideas an adjuvant therapy to PB in neonatal seizures
Clinical trials have investigated bumetanide as an adjunct, administered at a dose of 0.1-0.2mg/kgcon currently with PB to improve the efficacy of PB in neonates with PB-resistant seizures [30]. However, some concerns about bumetanide have been raised [31]. First, bumetanide functions as a potent loop diuretic with a short half-life in neonates [32]. In humans, NKCC2, another target of bumetanide, is robustly expressed in the kidney to extract and reabsorb ions from urine. Repeated doses of bumetanide can non-selectively block NKCC2 as well as NKCC1.Second, bumetanide has poor blood-brain barrier permeability that results in a less than 1% of bumetanide penetrating into the brain one hour after an intra peritoneal injection [33]. Although pathology-induced disruption of blood-brain barrier is expected in acquired neonatal seizures, the limited capability of bumetanide to reach the brain remains a major challenge [29]. Third, a few pre-clinical studies have reported that bumetanide is non-efficacious as an adjunct for anti-seizure pharmacotherapy [34,35]. One explanation for these findings is the possible KCC2 blockage by bumetanide in the seizing immature brain, which may result in an impaired extrusion of neuronal Cl-, further preventing GABAAR-targeted action of conventional anti-seizure agents. Finally, the NKCC1 knockout mouse is deaf, due to the loss of NKCC1 function in inner hair cells [36]. NKCC1 expression in the marginal cells of the inner ear is crucial for normal development of auditory function [36,37]. Therefore, systemic blockage of NKCC1 during the neonatal period could cause significant auditory deficits similar to those now evident in the NEMO study. The same isoform of NKCC1 is expressed in brain, hair cells, and kidney in humans [38], prohibiting alternative strategies like developing a CNS-specific NKCC1 blocker. This last concern became evident in the termination of the NEMO European clinical trial [NCT01434225], due to lack of anti-seizure efficacy of bumetanide associated with ototoxicity [15].
Model-specific variability of efficacy of bumetanide in preclinical studies of neonatal seizures
Understanding the injury-specific alterations of chloride co-transporters is a critical prerequisite for investigating pharmaco-modulation of chloride co-transporters [39]. Neuronal chloride gradients are not only affected by developmental shifts in co-transporter expression but also by the nature of the seizure-inducing injury. Altered function and expression of chloride co-transporters present one of the significant mechanisms for consequential seizure control, since chloride-co-transporters are key regulators of the neuronal Cl-gradient, which determine the efficacy of conventional anti-seizure agents. A few pre-clinical studies have reported the acute functional up regulation of NKCC1upon excitotoxic injury [40,41], which implicates a significant injury-induced alteration in the neuronal Cl-gradient even before the onset of any therapy. This acute post-injury up regulation of NKCC1 function may contribute to an increased Cl-concentration that may further prevent neuronal hyper polarization in response to GABAAR agonists. These pre-clinical observations originally led to the support and initiation of clinical trials on bumetanide as a promising adjuvant for treating refractory seizures in neonates.
Bumetanide has been studied in numerous pre-clinical models of neonatal seizures using chemoconvulsants [42,43], hypoxia [44], and ischemia [45], which all recapitulate the well-established neuronal hyperexcitability and higher seizure susceptibility during early development. However, the potential model-specificity in chloride co-transporter alteration following injury and its effect on the drug efficacy has been largely overlooked in recent reviews [31,46]. Chemoconvulsants, as shown in a kainic acid model, generate a substantial seizure-load that represent the status-like phenotypes frequently associated with HIE. However, chemoconvulsant-induced seizures may not be similar to ischemia- and hypoxia-induced seizures [Table 1]. Chemoconvulsant-related injury has been shown to up regulate KCC2 expression in contrast to an ischemia-related down regulation of KCC2, suggesting a differential modulation of KCC2 and NKCC1 by injury. Comparatively, hypoxic models, utilizing a hypoxia-only insult, consistent induction of transient seizures are reported; however the seizure loads are of a much lower severity compared to the chemoconvulsant and ischemia models. This low seizure-loads which consist of short duration seizures, may not result in excitotoxic injury similar to the other pre-clinical models nor represent the status-like seizure phenotypes associated with long-term morbidity and mortality in HIE [47].
![]()
Injury type
Model
Reference
Cl- co-transporter expression modulation on day of insult
Bumetanide
delivery paradigmBumetanide
anti-seizure efficacy at 0.1 - 0.2 mg/kg**Bumetanide with PB
anti-seizure efficacyChemo-convulsant
Kainic acid in P9 -12 rat
Dzhala et al., 20051
N/A
15 min
post-kainic acidAnti-seizure +
N/A
Kainic acid P5 -7 rat hippocampus
Khirug et al., 20102
KCC2 ↑NKCC1 N/A
1 h post-seizureN/A
N/A
N/A
Low Mg2+ in P4 - 6 rat
Dzhala et al., 20083
N/A
Post- 6 recurrent seizures induced by low Mg2+
Anti-seizure +
Anti-seizure +
PTZ in P7, 12, 18 rat
Mares., 20094
N/A
Pre-treatment
20 min prior to PTZNo effect
N/A
Hypoxia
15mins global hypoxia
in P10 ratCleary et al., 20135
KCC2 no change NKCC1 ↑� 1 h post-hypoxia
Pre-treatment
15 min prior to hypoxia onsetNo effect
Anti-seizure +
Ischemia
Permanent unilateral carotid ligation
in P7 and P10 mouse
Kadam et al., 20116
KCC2 ↓ NKCC1 ↑
4-24h post-ischemia (Fig. 1)2 h
post-ischemia onsetNo effect
No effect
Kindling
Rapid kindling for 80mins
in P11, 14, 21 rat
Mazarati et al., 20097
N/A
Pre-treatment
20 min prior to kindlingNo effect
N/A
Table 1: Pre-clinical models of electrographic neonatal seizures.
The mechanism by which the acute injury is induced seems to play a significant role in the modulation of chloride co-transporters, and ultimately in the translational value of the pre-clinical data. Acquired insults can differentially modulate the seizure susceptibility of the immature brain as well as the efficacy of pharmacotherapies being tested. Indeed, studies have reported different results on the post-injury alteration of chloride co-transporters, NKCC1 and KCC2 [33,45]. In a study utilizing a hypoxic injury, no change in the expression-levels of KCC2 and NKCC1were reported on the day of injury, in addition to a lack of anti-seizure efficacy of bumetanide at 0.15mg/kg on the same day [33]. In a study using an ischemic injury, a trend toward sup regulation of NKCC1 expression and significantly down regulated KCC2 expression was detected on the day of injury [45] and bumetanide showed no efficacy by itself or as an adjunct with PB. Similar NKCC1 up regulation after neonatal hypoxia ischemia have been reported previously [48]. This indicates that the underlying injury modulates chloride co-transporters expression differently in pre-clinical models of neonatal seizures; therefore the efficacy of agents that specifically modulate the function of chloride co-transporters, such as bumetanide, will also be different. This has indeed been observed and reported [49], explaining some of the model-specific variations in efficacy of bumetanide [29,50]. Hence, a critical review of models and model-specific alterations in chloride co-transporter expression and function may provide insight into differential effects of chloride co-transporter modulation and anti-seizure efficacy in pre-clinical, and their subsequent translational value.
KCC2 modulation after injury
The seizure-susceptible neuronal environment in the immature brain is well-established, but the pathology-related changes in KCC2 expression have not been consistently evaluated in pre-clinical models of neonatal seizures. Studies on the post-injury kinetics and expression-levels of KCC2 have been largely ignored, but one study using an adult is chemia has reported a significant acute post-injury downregulation in KCC2 protein [51,52] and mRNA level following interictal-like activity [53], which had not been reported in pre-clinical models of HIE-associated neonatal seizures. Ischemia can induce upregulation of Brain-Derived Neurotrophic Factor [BDNF] [54,55],which activates the Tyrosine receptor kinase B [TrkB] receptor signaling cascade that can lead to downregulation in KCC2 expression [51,56]. In vitro and in vivo studies on mature neurons have shown that BDNF upregulation results in a downregulation of KCC2 protein and mRNA expression in addition to suppressing its membrane-insertion and phosphorylation [53,57]. In contrast, in vitro studies on immature neurons reported that BDNF upregulation resulted in increased KCC2 protein expression [58,59]. KCC2 plays a pivotal role in Cl-homeostasis in the developmental context such that the shift of GABA action from excitatory to inhibitory may heavily depend on the age-dependent modulation of KCC2 function [60].
Summary
Translational research is best achieved when the model recapitulates the clinical condition as closely as possible with regards to mechanism of causation, severity, and progression of the disease. With the given caveat that animal models will never truly replicate the nuances and the wide variability expected in the patient population, the key question remains:“how well does the underlying pathology in the model truly represent the clinical condition?” Refractory seizures in neonates diagnosed with HIE are accompanied by additional complexities of energy failure and excitotoxic injury. Therefore, the mechanism by which injury is induced in a model may result in dissimilar activation of pathways, that may not reflect HIE pathophysiology. In the studies discussed above, the goal was to replicate neonatal seizures which clinically are known to be predominantly induced by HIE [7,8]. However, the model-specificity inherent to the mechanisms by which the seizures were induced highlights the differences in the efficacy of bumetanide as an anti-seizure drug. Dzhala et al. [23] used Chemoconvulsants to induce seizures; however, the effect of the chemoconvulsant insult on the expression of the targeted chloride co-transporters was not reported. Cleary et al. [33] utilized hypoxia to induce seizures, and showed stable KCC2expression at the acute time-point when bumetanide efficacy was tested, whereas our recent studies have demonstrated significant downregulation of KCC2 at acute time-points using an ischemic model (Figure 1). Not surprisingly, the efficacy of bumetanide as an anti-seizure agent is significantly different in all three models. In response to kainic acid-induced seizures [23], bumetanide showed a strong seizure suppression efficacy by itself. In contrast, pre-treatment with a similar dose of bumetanide in a hypoxia model showed no anti-seizure efficacy by itself on the mild hypoxic seizures [33]. In studies in our laboratory using a post-ischemia model that has a seizure severity similar to the status-like phenotypes reported in HIE [47], bumetanide had no anti-seizure effect even when delivered after PB (25mg/kg) as an adjunct treatment [45]. To understand these differences better, we investigated the modulation of chloride co-transporters after ischemia on day of insult and testing of BTN efficacy, and found that they were significantly different from each other [Figure 1]. Dzhala et al. did not report KCC2 expression levels after seizure induction, but significant KCC2 upregulation has been reported in studies using hippocampal slice cultures [61] derived from animals treated with Chemoconvulsants to induce seizures. These differences [Table 1] may underlie the differential efficacy of bumetanide reported in these pre-clinical models and discussed in previous reviews as a caution for the overall translational value of the bumetanide therapy [31,46]. Additionally, recent reviews of the literature in this field have now very eloquently discussed new and thought-provoking issues with regards to using bumetanide to treat refractory neonatal seizures [29,50,62].In spite of the valuable new insights offered, they do not address the injury-related differences in chloride co-transporter function in the pre-clinical models [63-65],which could explain the different efficacies in seizure suppression. In a few instances, protocols using pre-treatments with drugs that modify Cl-gradients even before the occurrence of an insult make it additionally difficult to interpret their translational value for a clinical condition in which the time point of insult is rarely known. Finally, an adverse benefit-risk ratio of bumetanide strongly advocates a consideration of alternative therapeutic strategies, like KCC2 modulation [29,62]. Recently, Glykys et al. [66] shed more light on the complexity of neuronal Cl-homeostasis, demonstrating its establishment not only by chloride co-transporters but also by local impermeant anions. It is not far-reaching to now hypothesize that these anions may also be differentially modulated in the different pre-clinical models of injury [Table 1]. Hence, it is crucial to ascertain how chloride co-transporters and the neuronal chloride gradient are modulated by the injury itself in pre-clinical models to attain additional insights into drug efficacies targeting those transporters and critical to investigate the same in patients with HIE.
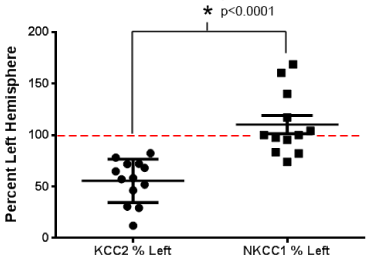
Figure 1 : Differentially modulatedKCC2 and NKCC1 expression in a mouse model of neonatal ischemia examined at 4-48h after ligation. Each data point represents an expression of KCC2 or NKCC1 of injured hemisphere of a mouse at postnatal day 7, normalized to the contra lateral side in a model of unilateral ischemic stroke. A significant downregulation of KCC2 expression compared to NKCC1 expression is the hallmark of the ischemia model.
Acknowledgement
Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R21HD073105 (SDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health; EFA Early Career Research Grant awarded to SDK (190631).
References
- Volpe JJ. Neonatal seizures. Clin Perinatol. 1977; 4: 43-63.
- Jensen FE. Neonatal seizures: an update on mechanisms and management. Clin Perinatol. 2009; 36: 881-900.
- Clancy RR. Summary proceedings from the neurology group on neonatal seizures. Pediatrics. 2006; 117: 23-27.
- Holden KR, Mellits ED, Freeman JM. Neonatal seizures. I. Correlation of prenatal and perinatal events with outcomes. Pediatrics. 1982; 70: 165-176.
- Ronen GM, Buckley D, Penney S, Streiner DL. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007; 69: 1816-1822.
- Glass HC, Pham TN, Danielsen B, Towner D, Glidden D, Wu YW. Antenatal and intrapartum risk factors for seizures in term newborns: a population-based study, California 1998-2002. J Pediatr. 2009; 154: 24-28.
- Tekgul H, Gauvreau K, Soul J, Murphy L, Robertson R, Stewart J, et al. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics. 2006; 117: 1270-1280.
- Sheth RD, Hobbs GR, Mullett M. Neonatal seizures: incidence, onset, and etiology by gestational age. J Perinatol. 1999; 19: 40-43.
- Sankar R, Painter MJ. Neonatal seizures: after all these years we still love what doesn't work. Neurology. 2005; 64: 776-777.
- Bartha AI, Shen J, Katz KH, Mischel RE, Yap KR, Ivacko JA, et al. Neonatal seizures: multicenter variability in current treatment practices. Pediatr Neurol. 2007; 37: 85-90.
- Garfinkle J, Sant'Anna GM, Wintermark P, Ali N, Morneault L, Koclas L, et al. Cooling in the real world: therapeutic hypothermia in hypoxic-ischemic encephalopathy. Eur J Paediatr Neurol. 2013; 17: 492-497.
- Low E, Boylan GB, Mathieson SR, Murray DM, Korotchikova I, Stevenson NJ, et al. Cooling and seizure burden in term neonates: an observational study. Arch Dis Child Fetal Neonatal Ed. 2012; 97: 267-272.
- Booth D, Evans DJ. Anticonvulsants for neonates with seizures. Cochrane Database Syst Rev. 2004.
- Pressler RM, Mangum B. Newly emerging therapies for neonatal seizures. Semin Fetal Neonatal Med. 2013; 18: 216-223.
- Pressler RM, Boylan GB, Blennow M, Chiron C, Cross JH, De Vries LS, et al. Bumetanide and hearing loss: results of a phase I/II dose finding & feasibility trial of bumetanide as add-on treatment for neonatal seizures: NEMO. 11th European Congress on Epileptology # p664; 2014.
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009; 5: 380-391.
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996; 16: 6414-6423.
- Dzhala VI, Staley KJ. Excitatory actions of endogenously released GABA contribute to initiation of ictal epileptiform activity in the developing hippocampus. J Neurosci. 2003; 23: 1840-1846.
- Lechpammer M, Manning SM, Samonte F, Nelligan J, Sabo E, Talos DM, et al. Minocycline treatment following hypoxic/ischaemic injury attenuates white matter injury in a rodent model of periventricular leucomalacia. Neuropathol Appl Neurobiol. 2008; 34: 379-393.
- González MI, Cruz Del Angel Y, Brooks-Kayal A. Down-regulation of gephyrin and GABAA receptor subunits during epileptogenesis in the CA1 region of hippocampus. Epilepsia. 2013; 54: 616-624.
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007; 87: 1215-1284.
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002; 3: 728-739.
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005; 11: 1205-1213.
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007; 160: 59-87.
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009; 61: 820-838.
- Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994; 269: 25677-25683.
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999; 274: 12656-12664.
- Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med. 2013; 19: 1524-1528.
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014; 55: 806-818.
- Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008; 63: 222-235.
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014; 55: 806-818.
- Pacifici GM. Clinical pharmacology of the loop diuretics furosemide and bumetanide in neonates and infants. Paediatr Drugs. 2012; 14: 233-246.
- Cleary RT, Sun H, Huynh T, Manning SM, Li Y, Rotenberg A, et al. Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLoS One. 2013; 8: 57148.
- Vargas E, Petrou S, Reid CA. Genetic and pharmacological modulation of giant depolarizing potentials in the neonatal hippocampus associates with increased seizure susceptibility. J Physiol. 2013; 591: 57-65.
- Mares P. Age- and dose-specific anticonvulsant action of bumetanide in immature rats. Physiol Res. 2009; 58: 927-930.
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999; 22: 192-195.
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, et al. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem. 1999; 274: 26946-26955.
- Delpire E, Kaplan MR, Plotkin MD, Hebert SC. The Na-(K)-Cl cotransporter family in the mammalian kidney: molecular identification and function(s). Nephrol Dial Transplant. 1996; 11: 1967-1973.
- Nardou R, Ferrari DC, Ben-Ari Y. Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med. 2013; 18: 175-184.
- Foroutan S, Brillault J, Forbush B, O'Donnell ME. Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl- cotransporter. Am J Physiol Cell Physiol. 2005; 289: 1492-1501.
- Dai Y, Tang J, Zhang JH. Role of Cl- in cerebral vascular tone and expression of Na+-K+-2Cl- co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol. 2005; 83: 767-773.
- Tremblay E, Nitecka L, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat. I. Clinical, electrographic and metabolic observations. Neuroscience. 1984; 13: 1051-1072.
- Khalilov I, Holmes GL, Ben-Ari Y. In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat Neurosci. 2003; 6: 1079-1085.
- Jensen FE, Applegate CD, Holtzman D, Belin TR, Burchfiel JL. Epileptogenic effect of hypoxia in the immature rodent brain. Ann Neurol. 1991; 29: 629-637.
- Kadam SD, Markowitz GJ, Kang SK, Kim ST, Johnston MV. Age and gender dependent severity of ischemic neonatal seizures: response to phenobarbital + bumetanide combination therapy in a mouse model. AES Annual meetings #3.035; 2011.
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014; 26: 34-41.
- Boylan GB, Stevenson NJ, Vanhatalo S. Monitoring neonatal seizures. Semin Fetal Neonatal Med. 2013; 18: 202-208.
- Dai Y, Tang J, Zhang JH. Role of Cl- in cerebral vascular tone and expression of Na+-K+-2Cl- co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol. 2005; 83: 767-773.
- Vanhatalo S, Hellström-Westas L, De Vries LS. Bumetanide for neonatal seizures: Based on evidence or enthusiasm? Epilepsia. 2009; 50: 1292-1293.
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013; 69: 62-74.
- Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010; 41: 151-159.
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008; 154: 677-689.
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004; 24: 4683-4691.
- Madinier A, Bertrand N, Mossiat C, Prigent-Tessier A, Beley A, Marie C, et al. Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One. 2009; 4: 8101.
- Béjot Y, Prigent-Tessier A, Cachia C, Giroud M, Mossiat C, Bertrand N, et al. Time-dependent contribution of non neuronal cells to BDNF production after ischemic stroke in rats. Neurochem Int. 2011; 58: 102-111.
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. 2002; 159: 747-752.
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010; 16: 302-307.
- Ludwig A, Uvarov P, Soni S, Thomas-Crusells J, Airaksinen MS, Rivera C. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J Neurosci. 2011; 31: 644-649.
- Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013; 2013: 429815.
- Dzhala VI, Kuchibhotla KV, Glykys JC, Kahle KT, Swiercz WB, Feng G, et al. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010; 30: 11745-11761.
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010; 30: 12028-12035.
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014; 26: 34-41.
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014; 55: 806-818.
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013; 69: 62-74.
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol. 2014; 26: 34-41.
- Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, et al. Local impermeant anions establish the neuronal chloride concentration. Science. 2014; 343: 670-675.