
Research Article
Austin J Cerebrovasc Dis & Stroke. 2024; 10(1): 1090.
ROS-Mediated Upregulation of MMP9 Expression via MAPK-AP1 Signaling Pathway and Disruption of Blood-Brain Barrier
Elizabeth Malcomson1,2; Wandong Zhang1,2*
¹Human Health Therapeutics Research Centre, National Research Council of Canada, Ottawa, Ontario, Canada
²Department of Cellular & Molecular Medicine, Faculty of Medicine, University of Ottawa, Ottawa, Ontario, Canada
*Corresponding author: Wandong Zhang Human Health Therapeutics Research Centre National Research Council of Canada, 1200 Montreal Road, Building M54 Ottawa, Ontario, K1A 0R6, Canada. Tel: 1-613-993-5988; Fax: 1-613-941-4475 Email: Wandong.Zhang@nrc-cnrc.gc.ca; wzhan2@uottawa.ca
Received: April 05, 2024 Accepted: May 06, 2024 Published: May 13, 2024
Abstract
Background: Ischemic stroke is associated with increased expression and activity of MMP9, which is one of the main factors responsible for damages to cerebral vasculature and compromised Blood-Brain Barrier (BBB). However, the regulatory mechanisms of MMP9 expression are not well established in ischemic stroke. Since ischemia/reperfusion generates ROS, we investigated the roles of Reactive Oxygen Species (ROS) in MMP9 expression and the relevant signaling pathway.
Methods: Cultured rat astrocytes were treated with ROS or/and MAPK inhibitors and the expression and activity of MMP9 were analyzed. AP-1 reporter gene assays and in vitro BBB model were also used in the study.
Results: Our study shows that ROS (H2O2) strongly up-regulated MMP9 expression in astrocytes via p38-and JNK-AP1 signaling pathways. MMP9 activity was significantly increased in the conditioned media from ROS-treated cells, leading to increased BBB permeability. The inhibitors of p38 kinase or JNK significantly inhibited ROS-evoked expression of MMP9 in cells and reduced MMP9 activity in the media. ROS also significantly activated AP-1 reporter gene activity. Inhibitors of p38 kinase and JNK inhibited ROS-evoked AP-1 reporter activity. In vitro BBB model assays show that inhibition of MMP9 expression and activity relieved increased BBB permeability.
Conclusion: Our study suggests that ROS can enhance MMP9 expression via p38 kinase-and JNK- AP1 pathways and promote MMP9 activity leading to increased BBB permeability and that inhibition of MMP9 activity can relieve BBB permeability. Targeting ROS or MMP9 may protect the integrity of the BBB for reduced damage and better recovery of ischemic stroke.
Keywords: Stroke; Hypoxia/ischemia and reperfusion; Reactive oxygen species; MMP-9; MAPK-AP1 signaling; Blood-brain barrier (BBB)
Introduction
Cerebral ischemia and reperfusion are known to result in oxidative stress and Blood-Brain Barrier (BBB) breakdown, which allows blood-borne substances to enter the brain and incites inflammatory response. Studies suggests that Matrix Metalloproteinase 9 (MMP9) can be activated by Reactive Oxygen Species (ROS) generated during ischemia and reperfusion and that ROS- induced up-regulation of MMP9 is one of the main factors responsible for degrading the Extracellular Matrix (ECM) proteins of cerebral vascular basal membrane and the tight junctions between cerebral endothelial cells leading to disruption of BBB integrity. Disruption of BBB integrity has dire consequences on the Central Nervous System (CNS), resulting in brain edema, infiltration of inflammatory cells, secondary brain damage and poor neurological outcomes [1,2]. MMP9 belongs to the family of Matrix Metalloproteinases (MMPs) which are dependent on metal ions for catalytic activity and their potent ability to degrade structural proteins of the extracellular matrix [3]. The MMPs exist as secreted or membrane-bound pro-enzymes that require activation through proteolytic processing from their pro-form (zymogen) to their active form [4]. Studies have recognized a role for MMPs in cortical changes of the brain following cerebral ischemia. MMP9 has been identified as being the most responsive MMP to acute brain injury [5]. MMP9 is capable of degrading type IV collagen, the major constituent of basement membranes of cerebral vessels. MMP9 is expressed in the healthy adult brain at relatively low levels [4]. However, MMP9 up-regulation occurs in neurons, astrocytes, oligodendrocytes, microglia and endothelial cells following acute brain injury. This level of MMP9 expression differs by the type (focal vs. global ischemia) [6] as well as by the duration and severity of the insult [4]. The up- regulation of MMP9 plays a deleterious role in the processes of extracellular proteolysis which contributes to tissue damage following acute brain injury [7], with the major pathological effect of MMP9 activity being the opening of the BBB compromising its integrity [4].
Oxidative stress/Reactive Oxygen Species (ROS) is generated during cerebral ischemia and reperfusion and plays a significant role in BBB disruption and brain edema. Previous studies in cell culture and animal models have indicated a free radical-induced oxidative stress as playing a significant role in ischemic brain injury [8-10]. More specifically, oxidative stress in cerebral ischemia and reperfusion can play a profound role in the disruption of the BBB [11]. Studies have shown that inhibition of MMP can prevent this oxidative stress-induced BBB disruption in ischemia [8,9]. The mechanism of action for ROS-mediated MMP activation was demonstrated in vitro by Rajagoplan et al [12], which showed ROS can oxidize a thiol bond responsible for activating MMP9. This suggests that macrophages activated during ischemia and reperfusion that release ROS and can trigger the activation of stored latent forms of MMP9 in the vascular regions of the BBB. Ashai and colleagues [7] showed that the inhibitor of ROS prevented tPA–triggered cerebral hemorrhage after stroke. In human patient’s oxidative stress has been shown to contribute to the pathogenesis of several neurodegenerative diseases and cerebrovascular disorders such as stroke and Alzheimer’s disease [13]. A human study evaluating biomarkers of antioxidant therapies in stroke shows that there was an increase in oxidative stress and this was related to MMP9 expression [14]. The aforementioned findings suggest that latent form MMP9 is activated at the protein level by oxidative stress and together these factors mediate BBB breakdown. Although MMP9 plays a significant role in the pathology of ischemic stroke, the signaling required for MMP- 9 up-regulation has yet to be fully elucidated in ischemic stroke. Since ROS is generated in ischemic stroke, it is important to understand whether these factors affect MMP-9 expression and its activity and the signaling pathways involved. Our study examines the molecular signaling mechanisms by which MMP9 becomes up-regulated by ROS-induced oxidative stress by using in vitro models whereby interference with this pathway could be developed as an approach to reduce MMP9-mediated BBB breakdown.
Materials and Methods
Cell Culture
Immortalized Neonatal Rat Astrocytes (NRA) were used in the experiments of this study [15,16]. NRAs were maintained in DMEM supplemented with 10% FBS and 1% antibiotic/antimycotic. The medium was changed every second day. HEK293 cells (human embryonic kidney epithelial cells) were maintained in DMEM with 10% FBS, and medium was changed every second day. HEK293 cells were plated at appropriate density for transfection protocol of the AP-1 reporter gene assays. Immortalized Rat Brain Microvascular Endothelial Cells (SV-ARBEC) were used in the in vitro model of the BBB for permeability assay [15,16]. SV- ARBEC cells were maintained in M199 medium supplemented with 10% FBS as described previously [15]. Cells were cultured on semi-permeable 1.0 μM pore membrane inserts that were coated with rat-tail collagen (60 μg/mL) for 1 h before plating. SV-ARBEC cells were grown for 6 days without feeding to allow a tight monolayer of cells to form.
RNA Isolation, RT-PCR, and Real-Time qPCR
NRA cells were treated with H2O2 (0.5, 1.0, and 2.0 μM) for 2, 4, and 6 h. Total RNA was isolated from cultured cells using TRIzol reagent following the manufacturer's instructions. RT- PCRs were performed as described [17]. All PCR primers (Table 1) were designed according to published sequences in the GenBank and synthesized by the Alpha DNA (Montreal, Quebec). For real-time quantitative PCR, RNA was converted to cDNA using Omniscript kit (Qiagen) according to manufacturers’ instructions. qPCR reactions were performed and analyzed as described [17].
Western Blot Analyses
Confluent NRA cells grown in 12-well culture plates were treated with 0.5, 1.0, and 2.0 μM H2O2 for 2, 4, and 6 h, respectively. Cells were harvested for proteins and Western blotting analysis was performed as described ([17]. At dilutions of 1:1000, primary antibodies of Phospho p38 (Thr180/Tyr182) MAP kinase mAb, p38 MAP kinase mAb, Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) mAb, p44/42 MAPK (ERK1/2) mAb, Phospho-c-Jun (Ser63) II mAb, Phospho- c-Jun (Ser73) II mAb and c-Jun (60A8) mAb (Cell Signaling) and Anti-MMP-9 Catalytic Domain mAb (Millipore) were used. Blots were developed using ECL Plus substrate solution according to manufacturer’s instructions and visualized using X-ray film.
Gelatin Zymography for MMP9 Activity in Astrocyte-Conditioned Media
Confluent NRA cells grown in 12-well culture plates were treated with 0.5, 1.0, and 2.0 μM H2O2 in the presence or absence of a pharmacologic inhibitor. After treatment, the conditioned media were collected, spun down by 10,000 rpm (9,000 g) centrifugation and mixed with equal amount non-reduced sample buffer (BioRad). Samples were loaded on a 10% gelatin SDS gel (precast by BioRad) and electrophoresed for 90 min at 100 V. Mixed blend of collagenases (Sigma) was used as a protein standard and positive control. Cell lysates were washed 1 time in colour-free HBSS and lysed in RIPA buffer for normalization with BioRad Protein Assay. BioRad protein assay was performed in 96-well plate where 2 mg/mL BSA was added in known amounts diluted with H2O providing a standard curve.
MMP9 Activity Assay
Confluent NRA cells in 24-well culture plates were pretreated with a pharmacologic inhibitor of MAP kinases at concentration of 20 μM for 30 min. After pretreatment, H2O2 was added to full culture media to final concentrations of 0.5 and 1.0 μM. H2O2 was left for 30 min, then media were switched for DME with 1% FBS for 2, 4, and 6 h. After treatment, conditioned media were collected, spun down by 10,000 rpm (9,000 g) centrifugation. Cell lysates were washed one time with colour-free HBSS and lysed in RIPA buffer for normalization with BioRad Protein Assay. MMP9-containing conditioned media were added to black 96-well plates. For every sample, 50 μL was added in triplicate and then 50 μL of warmed component D (Fret-peptide/ flourogenic substrate) was added. Plate was immediately placed into BioTek FLx800 Fluorescence Microplate Reader (Winooski, VT); readings were taken every 5 min for 1 h. Readings were normalized to 1% FBS DME media and reported as fold change in fluorescence.
Immuncytochemistry
NRA cells were plated at 80, 000 cells/mL in 24-well culture plates until reached 75% confluence. The cells were treated for 4 h with 1.0 μM H2O2 in regular culture media in the presence or absence of a pharmacologic inhibitor of MAP kinases at 20 μM. Media were aspirated; cells were washed 1 time in colour-free HBSS solution and then fixed by addition of ice-cold methanol for 5 min. Cells were washed in 4 times with HBSS. After ensuring integrity of the cells, they were permeablized with the addition of 0.1% Triton X-100 for 10 min at room temperature. Cells were washed in 3 times with HBSS for 5 min each. Cells were then blocked with 4% normal goat serum in HBSS for overnight at 4°C. Primary MMP9 antibody was then added at 1/100 dilution in 1% goat serum overnight at 4°C, and for secondary alone, control 1% goat serum in HBSS was added. Cells were washed in 2 times with HBSS before the addition of secondary Alexa568-conjugated anti-rabbit antibody at 1/500 dilution in HBSS, and the cells were incubated for 30 min at room temperature in the dark. Cells were then washed 3 times in HBSS before the addition of Hoechst 33342 at 1/5000 dilution in HBSS for 15 min. Images were taken with fluorescent microscope Olympus 1X81 (Center Valley, Pennsylvania) at 10x and 20x magnification.
Transient Transfection and AP-1 Reporter Gene Luciferase Assay
HEK293 cells were plated at 50,000 cell/well in 48-well plates or 100 000 cell/well in 24- well plates 24 h before transfection at which point they were 80 % confluent. HEK293 cells were transiently transfected with a reporter gene vector construct that contains AP-1 binding insert cloned from human MMP-9 gene promoter region into pGL-3 basic vector (Promega Corp.) using LipoFectamine transfection reagent (2:1 ratio of reagent to plasmid in μg). After 48 h recovery period at 37˚C, cells were treated with 1.0 μM PMA, 1.0 μM H2O2 or vehicle for 30 min, 2, 4, 6, and 24 h. The 30-min time point was determined to be the best for AP-1 activation by H2O2. After treatment cells were lysed in 1x cell lysis buffer provided in the Luciferase assay kit and spun down at 10,000 rpm for 2 min. Luciferase assay was performed using a kit purchase from Promega Co., (Cat# E1500, Madison, WI, USA). For luciferase assay, 20 μL of each sample was added to a black 96-well plate and 100 μL of luciferase enzyme was added by plate reader injector. Luminescence units were determined using BioTek FLx800 Fluorescence Microplate Reader (Winooski, VT). Luciferase units were normalized to protein in μg protein per sample using BioRad DC protein assay reagents previously described. HEK293 cells were used for reporter gene assay experiments due to low transfection efficiency in NRA cells (<5%).
In Vitro Blood-Brain Barrier Assay
An in vitro Blood-Brain Barrier (BBB) model was cultured using SV-ARBEC cells and conditioned media collected from confluent NRAs (15, 16). On day one of the experiment, cell culture inserts were coated with rat tail collagen at 60 μg/mL and SV-ARBEC cells were plated at the density of 80, 000 cells/mL. A 1:1 ratio of complete M199 phenol red free SV-ARBEC medium was mixed with DME phenol red free 1% FBS, NRA conditioned media (72 h off NRA confluent cells). Cells were incubated at 37°C for 6 days with no feeding for cells to form a tight monolayer. Before the experiment was done, the monolayer was tested for its permeability coefficient (Pe) with sodium fluorescein to ensure proper amount of in vitro transport across the in vitro BBB from the top chamber into the bottom chamber. In order to calculate Pe, culture inserts were gently lifted and washed 3X in warmed HBSS clear, and then placed in warmed 1X transport buffer. A known concentration of sodium fluorescein (50 μg/mL) was mixed 1:1 with pre-existing SV-ARBEC media, resulting in a final concentration of 25 μg/mL in the upper chamber. The plate was then placed into a shaking incubator at 37ºC. At time intervals of 15, 30, 45, 60, 90 and 120 min, 100 μL of transport buffer was removed from the bottom chamber, and 100 μL of fresh transport buffer was added. For inserts without SV-ARBEC cells, the same protocol was used with the exception of time intervals of 3, 7, 10, 15 and 20 min periodically during and after the experiment had been completed, the cell monolayers were checked under the microscope to ensure no folding or loss of cells indicating disrupted membrane integrity. After all of the time points had been collected, the 100 μL for each time point was added to a 96-well black plate and read in the plate reader at an excitation of 485 nm and an emission of 580 nm. After the Pe had been calculated as being between the standard rates for permeability generally between 0.5-0.8 X 10ˆ-3 cm/min, different treatments were added to the cells with a known concentration of sodium fluorescein and permeability was tested. The different treatments include: active MMP-9, active MMP-9 + MMP-9 inhibitor (SB- 3CT), H2O2 at 1.0 μM in NRA media, and conditioned media from NRA experiments where H2O2 had been treated for 4 h at 1.0 μM H2O2 and their respective vehicles. Standard curves were then used to calculate the amount of sodium fluorescein passing through the membrane, indicating the effect of treatments on BBB permeability. Once all time points were recorded, the permeability’s were compared to determine if treatment disrupted membrane integrity.
Statistical Analysis
Data were presented as mean ± SD. Statistical analysis for single comparison was performed by unpaired or paired Student’s t-test where each experiment was repeated at least 3 times (n=3). Statistical analysis for group comparison was performed by one-way ANOVA with Dunnett’s post-test where each experiment was done at least 3 times (n=3). The criterion for statistical significance was p<0.05. Statistical analysis for group comparison where two factors were being analyzed was performed by two-way ANOVA where each experiment was done at least 3 times with Bonferroni’s post-test (n=3). The criterion for statistical significance was p<0.05.
Results
H2O2-Induced Oxidative Stress Increased MMP9 Gene Expression in Astrocytes
Treatment of astrocytes (NRA cells) with H2O2 at 0.5, 1.0, and 2.0 μM for 2, 4, and 6 h was analyzed using semi-quantitative RT-PCR. Densitometry results showed a significant increase in the level of MMP9 mRNA in H2O2-treated NRAs. Gene expression was normalized using β-actin. The increase in gene expression is shown to peak with treatment of H2O2 at 1.0 μM for 4 h (data not shown). DME medium with H2O represented the vehicle internal control for H2O2 treatment and did not significantly affect MMP9 gene expression in NRAs. The RT-PCR result was confirmed by real-time qPCR, which confirmed 1.0 μM treatment for 4 h resulted in the greatest increase of MMP9 gene expression (Figure 1A). Gene expression was normalized using β-actin.
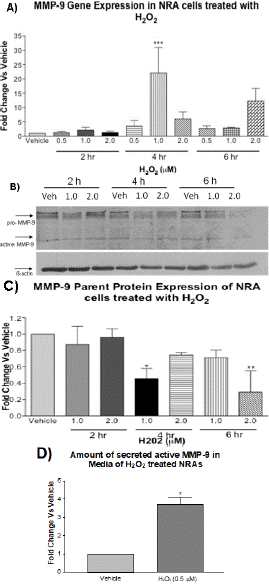
Figure 1: RT-qPCR, Western blotting, and MMP9 activity assays to determine the effects of H2O2 on MMP9 expression and activity in astrocytes. Panel A): RT-qPCR to determine MMP9 gene expression in NRA cells following treatment with H2O2 for 2, 4, and 6 h. Statistical analysis shows MMP9 expression was significantly higher in H2O2-treated cells at 4 h with 1.0 μM and 6 h with 2.0 μM as compared to vehicle (N=3, two-way ANOVA with Bonferroni’s post-test, interaction F6,24=4.12, *p<0.05, H2O2 treatment F3,24=4.37, *p<0.05 and time F2,24=4.89, *p<0.05). β-actin was used as an internal control. Panel B): Western blotting shows MMP9 protein expression in NRA cells following treatment with vehicle, 1μM H2O2, and 2μM H2O2 for 2, 4, and 6 h. Panel C): Densitometry analysis shows pro-MMP9 expression was significantly lower in H2O2-treated cells than vehicle at 4 h 1.0 μM and 6 h 2.0 μM (N=3, two-way ANOVA, with Bonferroni’s post-test, interaction F4,18=2.915, p>0.05, H2O2treatment F2,18=6.327, **p<0.001 and time F2,18=3.706, *p<0.05). β-actin was used for normalization. Panel D): MMP9 activity assay to determine the effect of H2O2 treatment on MMP9 activity in astrocyte-conditioned media. Conditioned media collected from astrocytes treated with vehicle and H2O2 at 0.5 μM in 1% FBS for 30 min followed by a medium switch for 1.5 h. MMP activity was measured as amount of cleaved fluorescent peptide as compared to vehicle (N=3, t-test, *p<0.001).
H2O2-Induced Oxidative Stress Decreased the Level of pro-MMP9 Protein in Astrocytes
To further understand the effect of H2O2 on MMP9 expression, western blotting was performed for the same treatments to determine the effect on pro-MMP9 expression. Figure 1B shows the treatment of NRA cells with H2O2 at 1.0 and 2.0 μM for 2, 4, and 6 h, and densitometry results show a significant decrease in the level of pro-MMP9 in H2O2-treated NRAs at 4 h and 6 h but not at 2 h (Figure 1C). Protein expression was normalized using β-actin. DME medium with H2O represents the vehicle internal control for H2O2 treatment and did not significantly affect MMP9 protein expression in NRAs. Western blot results demonstrated that H2O2-induced oxidative stress reduced the amount of pro-MMP9 with the greatest reduction occurring with treatment of 1.0 μM for 4 h and 2.0 μM 6 h. A decrease in MMP9 protein may be due to cleavage to the parent protein resulting in active form of MMP9.
H2O2-Induced Oxidative Stress Increased MMP9 Activity in Astrocyte-Conditioned Media
To show that this was in fact occurring, the conditioned media were tested on a sensitive MMP9 activity assay, to measure the activity of MMP9. The result showed a significant increase in active MMP9 in the media of 0.5 μM H2O2 treated NRA cells at 2 h (Figure 1D). In combination, PCR, Western blotting and activity assays show that MMP9 expression was significantly affected by H2O2 treatment.
Immunofluorescence of MMP9 to Detect Cellular Response to H2O2-Induced Oxidative Stress
To further understand the expression of MMP9 in NRA cells after H2O2-induced oxidative stress, immunocytochemistry was performed. NRA cells were treated with 1.0 μM H2O2 for 4 h to detect total MMP9 (Figure 2). Immunofluorescence with MMP9 antibody and Hoechst 33342 staining (nucleus) revealed a greater presence of MMP9 in NRA cells treated with H2O2 treatment than H2O alone. This result shows an overall increase in MMP9 in cells treated with H2O2. These results provide further insight into what was happening to MMP9 when the results of PCR, Western blot and MMP9 activity assays were considered as a whole picture that H2O2-induced oxidative stress resulted in an overall increase in MMP9 expression in astrocytes.
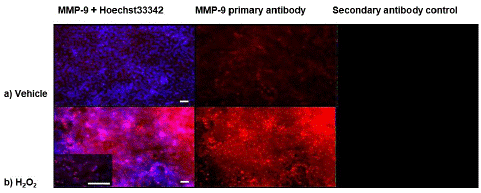
Figure 2: Immunoflourescence to detect the effect of H2O2 on the levels of MMP9 protein in astrocytes. Astrocytes were treated with 1.0 μM H2O2 for 4 h to detect total MMP9. Immunofluorescence with MMP9 antibody (red) and Hoechst 33342 staining (nucleus)(blue) revealed a greater presence of MMP9 in astrocytes treated with H2O2 treatment than H2O alone (N=3, images are representative). Secondary antibody control was done to detect non-specific binding. Images were captured at 10X magnification, higher power insert 20X magnification. Scale bars represent 50 microns.
H2O2-Induced Oxidative Stress Activated MAPK Signaling Pathways
To determine if Mitogen-Activated Protein Kinase (MAPK) signaling cascade is involved in oxidative stress-induced MMP9 expression in astrocytes, H2O2 treatment was repeated to detect the activation of the signaling proteins (phosphorylation) on MAPK pathways by Western blotting. Treatment of NRA cells with H2O2 at 0.5, 1.0, and 2.0 μM for 2, 4, and 6 h induced a significant increase in the phosphorylation of p38 kinase and c-Jun (Figure 3). Phosphorylation of p38 kinase increased in a dose-and time-dependent manner. Phosphorylation of c-Jun at Serine 63 and Serine 73 increased with concentration of H2O2 but levels peaked during time point of 4 h. Total levels of c-Jun protein did not change significantly with treatment of H2O2; however at 4 h time point the levels did trend upwards. In contrast, treatment of NRA cells with H2O2 at 1.0 and 2.0 μM for 2, 4, and 6 h induced a minimal change in phosphorylation of extracellular signal-regulated kinases 1/2 (ERK1/2) (data not shown). All phosphorylation levels were normalized using β-actin. DME medium with H2O represents the vehicle internal control for H2O2 treatment and did not significantly affect phosphorylation levels of MAP kinase proteins in NRAs. The phosphorylation data suggests that the MAP kinase signaling pathways p38 and JNK may be highly involved in signaling during oxidative stress in NRA cells. By comparison, the ERK pathway may not be playing as important of a role. The MAP kinase phosphorylation profiles with H2O2 treatment are in line with previous research that suggests ROS can activate the JNK, p38, and ERK pathways. Generally environmental stressors activate JNK and p38 kinases whereas growth factors and tumor promoters activate ERK1/2 [17]. The MAPK phosphorylation results provide the insight that p38, JNK, and ERK1/2 known activators of AP-1, could potentially be upstream regulators of MMP9 expression.
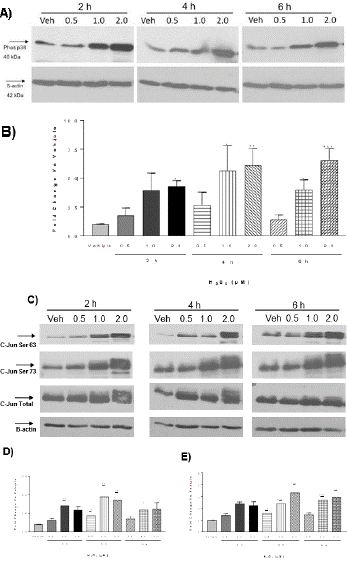
Figure 3: Western blotting to detect phosphorylation of p38 kinase and c-Jun in astrocytes in response to H2O2-induced oxidative stress. Panel A). Western blotting shows the level of phosphorylated p38 kinase in astrocytes following treatment with H2O2 for 2, 4, and 6 h. Panel B). Densitometry analysis shows phosphorylated p38 was significantly greater in H2O2-treated cells 2.0 μM at 2 h, 1.0 and 2.0 μM at 4 h, and 1.0 and 2.0 μM at 6 h as compared to vehicle (N=3, Two-way ANOVA with Bonferroni’s post-test, Interaction F6,24 = 0.4979, p>0.05; H2O2 treatment F3,24=13.31, *p<0.05, **p<0.01; Time F2,24=1.163, p>0.05). ß-actin was used for normalization. Panel C). Western blotting shows the level of phosphorylated c-Jun at Serine 63, the level of phosphorylated c-Jun at Serine 73 and the total c-Jun in NRA cells following treatment with vehicle and H2O2 at 0.5, 1.0, and 2.0 μM for 2, 4, and 6 h. Panel D). Densitometry analysis shows the level of phosphorylated c-Jun at serine 63 was significantly greater in H2O2-treated cells than vehicle at 4 h (N=4, Two-way ANOVA with Bonferroni’s post-test, Interaction F6,33=0.5704, p>0.05; H2O2 treatment, F3,33=7.452, *p<0.05, **p<0.01, *** p<0.001; Time F2,33=1.409, p>0.05). β-actin was used for normalization. Panel E). Densitometry analysis shows phosphorylated c-Jun at serine 73 was significantly greater in H2O2-treated cells than vehicle at 2, 4, and 6 h (N=3, Two-way ANOVA with Bonferroni’s post-test, Interaction F6,36=0.6247, p>0.05; H2O2 treatment F3,36=18.38, *p<0.05, **p<0.01, ***p<0.001; Time F2,33=1.409, p>0.05). -actin was used for normalization.
Characterization of the Roles of MAPK Signaling Pathways in Regulation of MMP9 Expression
Results of phosphorylation of MAP kinases by western blotting suggested a potential role of MAP kinases in regulation of MMP9 expression. To determine if MAP kinases were playing a role in regulation of MMP9 expression, the levels of MMP9 mRNA, protein and MMP9 activity were studied in the presence of chemical inhibitors specific to JNK, p38 and ERK1/2. To determine if the inhibitors of MAP kinases could reduce mRNA levels of MMP9, the time points of H2O2 treatment from previous experiments that yielded greatest increase (1.0 μM for 4 h) were used. NRA cells pre-treated for 30 min with chemical inhibitors for p38 (SB203580), JNK (SP60015), and ERK1/2 (U0126) at 10 and 20 μM showed a significant reduction in MMP9 mRNA levels by qRT-PCR (Figure 4A). This result suggests that in NRA cells the MAP kinase signaling pathways are involved in up-regulation of MMP9 expression following H2O2-induced oxidative stress.
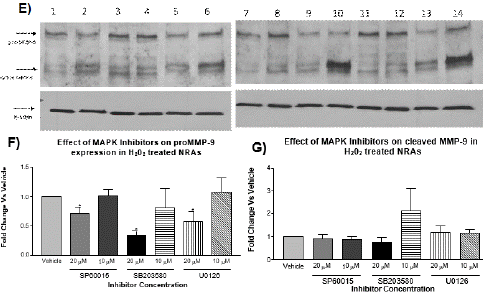
Figure 4: RT-qPCR and Western blotting for MMP9 gene expression in astrocytes in the presence of MAPK inhibitors. Panel A): Astrocytes were pre-treated with MAP kinase inhibitors SP60015 (JNK inhibitor), SB 203580 (p38 kinase inhibitor), and U0126 (ERK1/2 inhibitor) for 30 min at 10 and 20 μM before treating with H2O2 at 1.0 μM for 4 h. Treatment with MAP kinase inhibitors reduced the relative expression of MMP9 by real-time qPCR as compared to vehicle (N=3, two-way ANOVA with Bonferroni’s post-test, Interaction F3,16=0.3538, p>0.05; MAPK inhibitor F3,16=13.52, *p<0.05,**p<0.01 ,***p<0.001; Concentration F1,16=0.00052, p>0.05). β-actin was used for normalization. Panel B): Western blotting for astrocytes pre-treated with 0.1% DMSO for 30 min before treating with vehicle-H2O or H2O2 at 1.0 μM for 6 h. Panel C): Densitometry analysis shows pro-MMP9 expression was not affected by the presence of 0.1% DMSO and was significantly lower in H2O2 -treated cells than vehicle at 6 h (N=3, t test, *p<0.05). β-actin was used for normalization. Panel D): Densitometry analysis shows cleaved form of MMP9 expression was significantly higher in H2O2-treated cells than vehicle at 6 h (N=3, t test, *p<0.05). β-actin was used for normalization. Panel E): Western blotting for MMP9 shows astrocytes pre- treated with MAP kinase inhibitor SP60015 (JNK inhibitor), SB 203580 (p38 inhibitor) or U0126 (ERK1/2 inhibitor) for 30 min at 10 and 20 μM before treating with H2O2 at 1.0 μM for 6 h. Lane #1: vehicle (0.1% DMSO); Lane #2: vehicle + H2O2 (1μM); Lane #3: JNK inhibitor (20μM); Lane #4: JNK inhibitor (10μM); Lane #5: JNK inhibitor (20μM) + H2O2 (1μM); Lane #6: JNK inhibitor (10μM) + H2O2 (1μM); Lane #7: p38 inhibitor (20μM); Lane #8: p38 inhibitor (10μM); Lane #9: p38 inhibitor (20μM) + H2O2 (1μM); Lane #10: p38 inhibitor (10μM) + H2O2 (1μM); Lane #11: ERK inhibitor (20μM); Lane #12: ERK inhibitor (10μM); Lane #13: ERK inhibitor (20μM) + H2O2 (1μM); and Lane #14: ERK inhibitor (10μM) + H2O2 (1μM). Panel F): Densitometry analysis of Western blotting for pro-MMP9 (including the Western blot in Panel E) shows that treatment with MAP kinase inhibitors at 20 μM significantly reduced the expression of pro-MMP9 (92 kDa) as compared to vehicle (N=3, one-way ANOVA with Dunnett’s post-test, F3,8=6.519, *p<0.05). -actin was used for normalization. Panel G). Densitometry analysis of Western blotting for cleaved MMP9 (including the Western blot in Panel E) shows that treatment with MAP kinase inhibitors had no significant effect on the levels of cleaved-MMP9 as compared to vehicle (N=3, one-way ANOVA, p>0.05).
Western blotting was then done for pro-MMP9 and cleaved MMP9 (active form) in cells treated with 1.0 μM for 4 and 6 h. The vehicle controls demonstrated that H2O2-induced oxidative stress decreased the amount of parent protein but increased the amount of cleaved active MMP9 protein. This further validates the previous result that although parent protein was reduced due to cleavage, the cleaved amount of MMP9 was increased (Figure 4B). Treatment with JNK, p38 and ERK inhibitor (20 μM) further reduced the amount of pro-MMP9 parent protein at 6 h without significant effect on cleaved MMP9 active protein (Figure 4C). Together, these results demonstrate that the MAP kinase signaling cascade plays a role in the regulation of MMP9 expression. Gene and protein expression data demonstrates that inhibition of MMP-9 expression is achieved by blocking MAP kinase signaling pathways.
To further understand the effect of MAP kinases on the expression of MMP9 in NRA cells after H2O2-induced oxidative stress, immunocytochemistry was performed. NRA cells were pre- treated with JNK, p38 or ERK inhibitor (20μM) for 30 min before treatment with 1.0 μM H2O2 for 4 h to detect the levels of cellular MMP9 (Figure 4F & 4G). Immunofluorescence with a MMP9 antibody and Hoechst 33342 staining revealed a reduction of MMP9 in NRA cells treated with the inhibitors + H2O2 treatment compared to controls (Figure 5). These results show an overall decrease in MMP9 in cells treated with JNK and p38 inhibitor + H2O2, consistent with previous PCR, western blot and MMP activity assay results. This suggests that JNK and p38 kinases play a significant role in the signaling of MMP9 expression in H2O2-induced oxidative stress.
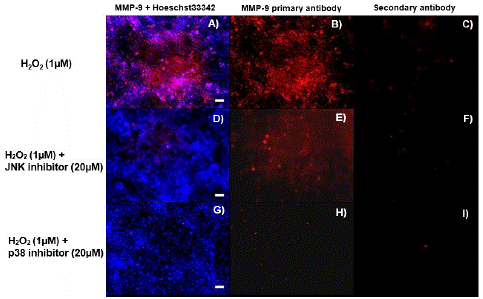
Figure 5: Immunofluorescence to detect the levels of MMP9 protein in astrocytes pre-treated with MAPK inhibitors. Immunofluorescence shows MMP9 protein in astrocytes pre-treated with MAP kinase inhibitor SP60015 (JNK inhibitor) or SB 203580 (p38 inhibitor) for 30 min at 20 μM before treating with H2O2 at 1.0 μM for 4 h. The images are: A) astrocytes treated with H2O2 and stained with MMP9 antibody (red) and Hoechst33342 (blue); B) astrocytes treated with H2O2 and stained with MMP9 antibody; C) astrocytes treated with H2O2 and stained with secondary antibody only; D) astrocytes treated with SP60015 (JNK inhibitor) + H2O2 and stained with MMP9 antibody and Hoechst33342; E) astrocytes treated with SP60015 (JNK inhibitor) + H2O2 and stained with MMP9 antibody; F) astrocytes treated with SP60015 (JNK inhibitor) + H2O2 and stained with secondary antibody only; G) astrocytes treated with SB 203580 (p38 inhibitor) and H2O2 and stained with MMP9 antibody and Hoechst33342; H) astrocytes treated with SB 203580 (p38 inhibitor) and H2O2 and stained with MMP9 antibody; and I) astrocytes treated with SB 203580 (p38 inhibitor) and H2O2 and stained with secondary antibody only. Images were captured at 10X magnification. Scale bar represents 50 microns.
MAPK Signaling Pathway Mediates MMP9 Expression Through AP-1.
HEK293 cells were transiently transfected with pGL-3 reporter gene vector, which carries an insert from AP-1 binding site cloned from the promoter region of human MMP-9 gene [17]. In addition, HEK293 cells were also transiently transfected with a commercial pTL-Luc reporter gene vector that contains multiple repeats of AP-1 binding site [17]. The controls for the assay were the transfection of cells with an empty pGL-3 or pTL vector without the AP-1 insert. After 48 h recovery post-transfection, the cells were treated in the presence of 20 μM MAPK inhibitors for 30 min followed by 1.0 μM H2O2 for 30 min. PMA was used as a positive control known to induce AP-1 activity in HEK293 cells. Reporter gene assays show the effect of H2O2 on AP-1 induction and the MAPK inhibitors on reducing AP-1 induction, since AP-1 induction was directly proportional to luciferase activity. Data shows a significant AP-1 induction in H2O2-stimulated HEK293 cells (Figure. 6A & 6B). This result indicates that H2O2-induced MMP9 gene expression goes through the MAPK-AP1 pathway, suggesting that ROS activates the MAP kinases, and the activated MAP kinases activate AP-1 which then turns on MMP9 gene expression. The MAPK inhibitors were not all able to reduce AP-1 reporter gene activity in the commercial AP-1 vector but all reduced the activity of AP-1 in the MMP9 promoter vector (Figure 6C & 6D).
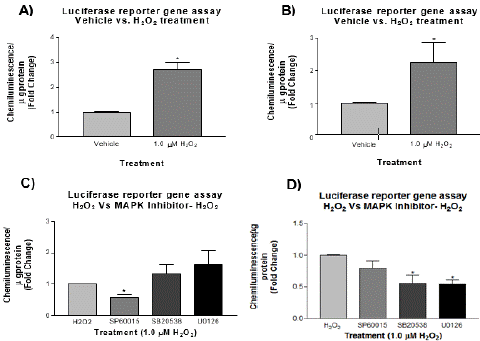
Figure 6: Reporter gene assay for the effect of H2O2 on AP-1 transcriptional activity. HEK293 cells were transfected with pTL/AP-Luc vector (Panel A) and pGL-3/AP1 (Panel B) vector and then treated with H2O2 at 1.0 μM for 30 min. H2O2 strongly stimulated reporter gene activity of pTL/AP-Luc plasmid at 30 min (N=3, t-test, *p<0.05). pGL-3/AP-1 plasmid showed the similar response at 30 min (N=3, t-test, one-tailed p<0.05). HEK293 cells were transfected with pTL/AP- Luc vector (Panel C) or pGL-3/AP-1 vector (Panel D) and then pretreated with MAP kinase inhibitors, SP60015 (JNK inhibitor), SB 203580 (p38 inhibitor) or U0126 (ERK1/2 inhibitor) for 30 min at 20 μM before treatment with H2O2 at 1.0 μM for 30 min. Pre-treatment with SP60015 (JNK inhibitor) in pTL/AP-Luc vector significantly reduced the reporter gene activity (Panel C: N=3, One-way ANOVA, p>0.05, t-test, *p<0.05). Pre-treatment with all MAPK inhibitors in pGL- 3/AP-1 vector reduced reporter gene activity (Panel D: N=3, one-way ANOVA with Dunnett’s post test F3 8=6.152 *p<0.05).
Inhibition of MMP9 Expression Reduced the Permeability of an In Vitro BBB Model
A two-chamber in vitro model of the BBB using SV-ARBEC cells was used to measure barrier permeability with the fluorescent dye marker sodium fluorescein as described [15]. Initially, Pe test was used to ensure that the brain endothelial cells form a tight monolayer on the semi- permeable membrane of the culture inserts. Three wells with the cells were compared to three wells without cells (empty wells) to determine the permeability in a top-down direction of the cell layer. Pe test demonstrates that after 6 days in culture, the permeability coefficient of the in vitro BBB model was 0.56 X 10-3 cm/min for top to bottom transport (Figure 7A). The Pe value indicates that the BBB monolayer was tight and the permeability of the in vitro BBB model was low. Previous studies suggest ROS such as H2O2 is involved in the disruption of the BBB during cerebral ischemia or ischemic stroke. Thus, the in vitro model was tested whether H2O2 does have this effect. The treatment of the in vitro BBB model with 1.0 μM H2O2 showed a similar effect, in that the permeability or leakage of the BBB was increased in the presence of ROS (Figure 7B).
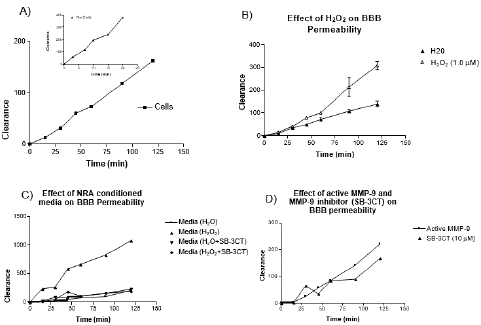
Figure 7: Effect of H2O2 and MMP9 expression on the transport of fluorescein across an in vitro BBB model. The in vitro BBB model consists of SV-ARBEC cells grown as a monolayer on a porous membrane in the tissue culture insert. Media conditioned by rat astrocytes were applied to the bottom compartment to induce the BBB phenotype. Fluorescein (25 μg/mL) was added to the top compartment and concentrations were determined in the bottom compartment by fluorescence detection. Panel A): Permeability test was conducted determining the fluorescein concentration across triplicate membranes with and without cells. Panel B): H2O2 at concentration of 1.0 μM increases monolayer permeability and fluorescein transport across the membrane. Panel C): Conditioned media collected from NRA cells treated with H2O2 at concentration of 1.0 μM increased monolayer permeability and fluorescein transport across the membrane but treatment with MMP9 inhibitor SB-3CT at 10 μM concentration maintained monolayer integrity and reduced fluorescein transport. Panel D): Active MMP9 (25 units/mL) increased monolayer permeability and fluorescein transport across the membrane but treatment with MMP9 inhibitor SB-3CT at 10 μM concentration maintained monolayer integrity and reduced fluorescein transport.
We tested if conditioned media from NRA cells treated with the same concentration of H2O2 could have a similar effect or an even greater effect due to the presence of potentially active MMP9 produced or released from the cell. The results suggest that in fact conditioned media was capable of greatly increasing the BBB permeability. To determine if this permeability was due a presence of active MMP9 in the conditioned media, MMP9 inhibitor SB-3CT was added to the media. The MMP9 inhibitor was capable of reducing BBB leakage (Figure 7C). ROS was capable of increasing BBB permeability by activating latent forms of MMP-9 and increasing MMP-9 expression. This led us to test whether recombinant active MMP-9 could have a similar effect on BBB permeability. The result shows there was increase in permeability in the presence of active MMP9 and that the addition of MMP9 inhibitor SB-3CT was able to reduce this effect (Figure 7D).
Discussion and Conclusion
This work is to understand the signaling mechanisms that regulate ROS-evoked MMP9 expression, leading to BBB disruption due to ischemic stroke by using in vitro models. The physical barrier of the BBB is made up of tight junctions between endothelial cells that restrict the passage of solutes between the blood and brain [19]. These tight junctions are built upon the endothelial cell matrix made up of collagens, laminin and fibronectin. Collagen type IV has been shown to directly influence the expression of occludin at tight junctions [20]. Loss of laminin, collagen and fibronectin can disrupt the endothelial tight junctions and loss of BBB integrity [21,22]. ECM degradation and disruption of basal lamina is the primary cause of microvascular hemorrhage following an ischemic event. Increased MMP9 expression was correlated with BBB leakage in animal models of cerebral ischemia [23]. Elevated MMP9 levels coincide with neurological outcome, suggesting that MMP9 plays a role in neuronal death and brain injury following ischemic stroke [24]. Knockout study has demonstrated that the MMP9 deficient mice were protected against brain trauma and focal cerebral ischemia [25] and show reduced BBB leakage [7]. MMP9 inhibitors were shown to prevent laminin degradation, which maintains ECM and BBB integrity and protects neurons from apoptosis [26,27]. Thus, increased levels of MMP9 following acute brain ischemic injury have been linked to greater infarct size, poor neurological outcome, and hemorrhagic transformation complications [28]. MMP9 may be a potential pharmacological target and biomarker of stroke, as high levels of this protease were detected in the blood of human patients following ischemic and hemorrhagic strokes [24].
MMP9 plays a significant role in the pathology of ischemic stroke, whereby it aides in the detrimental breakdown of the BBB; however, the signaling required for MMP9 up-regulation still remains largely uncharacterized in ischemic stroke. A recent study shows that MMP9 expression in androgen receptor-positive triple-negative breast cancer cells is regulated epigenetically via AKT/mTOR signaling and suppressed by luteolin [29]. Luteolin was also used as a treatment option of ischemic stroke in animal MCAO model by inhibiting MMP9 and activation of the PI3K/Akt signaling pathway [30]. Another study shows that lysophosphatidic acid promotes expression and activation of MMP9 in THP-1 cells via TLR4/NF-κB signaling pathway [31]. In this study we demonstrate that H2O2-induced oxidative stress stimulated a profound change in the expression of MMP9 in astrocytes. The response to H2O2 involves activation of p38, JNK and ERK signaling pathways that converge onto AP-1. Furthermore, our work with the in vitro BBB model suggests that active MMP9 and H2O2 both increased the permeability of the BBB demonstrating their potential as therapeutic targets. Gene expression work demonstrated that H2O2 significantly induced MMP9 expression at mRNA level. Interestingly, protein expression data was not as easily understood as Western blot results showed that H2O2 decreased the amount of pro-MMP9 in astrocytes. It at first seems unlikely that gene expression at the mRNA level was increased where protein was decreased. Since the experiment was not effectively measuring total MMP9 in astrocytes and the cleaved active form of MMP9 released into the culture media, MMP9 activity assay was used and showed that the amount of active MMP9 in the conditioned media of astrocytes was significantly greater than controls. This suggested that in fact although the amount of MMP9 was increased in astrocytes, immunocytochemistry confirmed that overall amounts MMP9 were remarkably increased in the astrocytes treated with H2O2. These findings demonstrate that in fact oxidative stress can induce MMP9 expression at many different levels and ROS may serve as a key-signaling player in the activation of MMP9 expression and the consequential BBB damage.
Gashe and colleagues showed that MMP inhibition prevented the oxidative stress- associated BBB disruption following cerebral ischemia and suggested that oxidative stress mediates BBB disruption by activation of MMPs [9]. Macrophages activated during cardiovascular injury release ROS that activate MMP2 and MMP9, which then can degrade the collagen components of ECM resulting in negative outcomes for patients [32]. The mechanism of action for ROS-mediated MMP activation is that in vitro ROS can oxidize a thiol bond responsible for activating MMP2 and MMP9. This suggests that macrophages activated during ischemia and reperfusion that release ROS can trigger the activation of stored latent forms of MMP9 in the vascular regions of the BBB [12]. Our work adds to this body of literature indicating that ROS produced by oxidative stress during cerebral ischemia/reperfusion can induce the expression and activate MMP9. We investigated the involvement of p38, JNK and ERK1/2 signaling during H2O2-induced MMP9 expression. Protein work demonstrated that phosphorylation levels of p38 and JNK kinase were greatly increased with the ROS treatment but not the ERK 1/2 kinase. Previous research on the MAPK signaling pathways has shown that the same types of stimuli generally affect p38 and JNK kinase; while ERK1/2 is activated by growth factors and tumor promoters. The phosphorylation profiles suggest that the MAP kinase signaling cascades could be the upstream activators of MMP9 gene expression, specifically p38 and JNK. Chemically specific inhibitors of MAP kinases were then used to determine the effect of blocking these pathways would have on gene and protein expression. As expected, the inhibitors were able to significantly reduce the gene expression and further decrease the protein expression of MMP9. All three inhibitors are involved, to some degree, in inhibiting the signaling of MMP9, as no one inhibitor was capable of completely abolishing the expression, but were capable of significantly reducing it. This work clarifies what was occurring with H2O2 treatment and MMP9 gene expression, revealing that although the pro- MMP9 was decreased, the cleaved MMP9 (active MMP9) was increased significantly with the treatment. However, the chemical inhibitors were not capable of significantly reducing the amount of cleaved MMP9, indicating that the inhibitors cannot inhibit H2O2 ability to cleave the existent parent MMP-9 (pro-MMP9) within cells. Therefore, it is important to note that the treatment with these inhibitors reduced overall MMP9 expression. This was further demonstrated by immunocytochemistry work revealing that JNK and p38 inhibitors reduced MMP-9 expression. It is of interest to note that although ERK inhibitor was capable of reducing MMP-9 expression at the mRNA level, it did not reduce overall MMP9 expression in cells and ERK was not phosphorylated by H2O2 stimulation as observed for p38 and JNK. It is likely that perhaps ERK 1/2 is involved in MMP-9 signaling in normal physiological processes not in response to stressors. The last aspect of characterizing the signaling pathways for MMP-9 up-regulation is the transcription factor that activates transcription of the gene of interest. The MAP kinase signaling pathways are well conserved and composed of a well-studied family of kinases known to converge on AP-1 transcription factor. Thus, we examined a link between H2O2 treatment and AP-1 activation. The reporter gene assays with the vectors containing the two upstream AP-1 binding sites from human MMP-9 and the classic AP-1 binding sites showed significantly greater luciferase activity, indicating that H2O2 could influence MMP9 gene expression through AP-1. All three MAP kinases inhibitors reduced the reporter gene activity. This confirms that in fact MAP kinases can activate MMP9 gene expression to some degree through AP-1. The signaling pathway research demonstrated the complexity of MMP-9 up-regulation in brain cells. Characterization of MMP9 is difficult due to its varying forms and the ability of molecules such as ROS to both activate and latent forms of MMP9 as well as to induce signaling for generation of MMP9. In addition, the three MAP kinase-signaling pathways have been shown in this research to play to some extent a role in MMP9 signaling. This suggests that although one signaling pathway may be inhibited, there could be still generation and compensation by other pathways. Given the abovementioned results, inhibition of MAPK signaling may decrease the expression of MMP9 and relieve oxidative stress-induced MMP9 expression and consequential BBB damage. Our results suggest that both H2O2 and MMP9 play a role in disrupting monolayer integrity of the in vitro BBB model. Treatment with MMP9 inhibitor was able to reduce the monolayer damage caused by astrocyte-conditioned media containing H2O2 and MMP9. Our work provides the insight into what treatments may be effective in preventing MMP9-mediated BBB breakdown in ischemic stroke.
Author Statements
Acknowledgement
The authors would like to thank Dr. Danica Stanimirovic at the National Research Council of Canada (NRC) for providing the immortalized rat astrocytes and brain endothelial cells for the study and Ms. Debbie Callaghan and Ms. Joy Lei at NRC for their assistance.
Funding
The work was supported by a research grant from the Heart & Stroke Foundation of Ontario (#NA6473) to Dr. W. Zhang. The study was conducted at the Human Health Therapeutics Research Centre, National Research Council of Canada.
Author Contributions
EM designed and conducted the experiments, performed data analyses, drafted the figures and the manuscript. WZ conceived the idea, designed the experiments, performed some data analyses, drafted and revised the manuscript.
Data Availability Statement
Data is available upon request.
References
- Rosell A, Ortega-Axnar A, Alvarez-Sabin J, Fernandez-Cadenas I, Ribo M, Molina CA, et al. Increased brain expression of matrix metalloproteinase-9 after ischemia and hemorrhagic human stroke. Stroke. 2006; 37: 1399-1406.
- Zlokovic BV. Remodeling after stroke. Nat Med. 2006; 12: 390-391.
- Stocker W, Bode W. Structural features of the superfamily of zinc-endopeptidases: the metzincins. Curr Opin Struct Biol. 1995; 5: 383-390.
- Cunningham L, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 2005; 50: 329-339.
- Lee SR, Kim HY, Rogowska J, Zhao BQ, Bhide P, Parent JM, et al. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J. Neurosci 2006; 13: 3491-3495.
- Rosenberg GA, Mun-Bryce S. Matrix metalloproteinases in neuroinflammation and cerebral ischemia. Ernst Schering Res Found Workshop. 2004: 1-16.
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001; 19: 7724-7732.
- Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, et al. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997; 17: 4180-4189.
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PK. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain-barrier disruption after transient focal cerebral ischemia. J. Cereb Blood Flow & Metab. 2001; 21: 1393-1400.
- Kamada H, Yu F, Nito C, Chan PH. Influence of Hyperglycemia on Oxidative Stress and Matrix Metalloproteinase-9 Activation After Focal Cerebral Ischemia/Reperfusion in Rats. Stroke. 2007; 38: 1044-1049.
- Shukla A, Shukla R, Dikshit M, Srimal RC. Alterations in free radical scavenging mechanisms following blood-brain barrier disruption. Free Radic Biol Med. 1993; 15: 97-100.
- Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive Oxygen Species Produced by Macrophage-derived Foam Cells Regulate the Activity of Vascular Matrix Metalloproteinases In Vitro Implications for Atherosclerotic Plaque Stability. J. Clin Invest. 1996; 98: 2572–2579.
- Uno M, Harada M, Takimoto O, Kitazato KT, Suzue A, Yoneda K, et al. Elevation of plasma oxidized LDL in acute stroke patients is associated with ischemic lesions depicted by DWI and predictive of infarct enlargement. Neurol Res. 2005; 27: 94-102.
- Kelly PJ, Morrow JD, Ning MM, Koroshetz W, Lo EH, Terry E, et al. Oxidative Stress and Matrix Metalloproteinase-9 in Acute Ischemic Stroke. Stroke. 2008; 39: 100-104.
- Zhang W, Mojsilovic-Petrovic J, Andrade M, Zhang H, Ball M, Stanimirovic D. Expression and Functional Characterization of ABCG2 in Brain Endothelial Cells and Vessels. FASEB J. 2003; 17: 2085-2087.
- Garberg P, Ball M, Borg N, Cecchelli R, Fenart L, Hurst RD, et al. In vitro models for the blood-brain barrier. Toxicol In Vitro. 2005; 19: 299-334.
- Vukic V, Callaghan D, Walker D, Lue L-H, Liu Q-Y, Stanimirovic DB, et al. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009; 34: 95-106.
- Simon C, Simon M, Vucelic G, Hicks MJ, Plinkert PK, Koitschev A, et al. The p38 SAPK Pathway Regulates the Expression of the MMP-9 Collagenase via AP-1 Dependant Promoter Activation. Exp Cell Res. 2001; 271:344-355.
- Terstappen G, Meyer A, Bell R, Zhang W. Strategies for delivering central nervous system therapeutics across the blood-brain barrier. Nature Review Drug Discovery. 2021; 20: 362-383.
- Savettieri G, Liegro I, Catania C, Licata L. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuro Report. 2000; 11: 1081-1084.
- Hamann GF, Okada Y, Fitridge R, Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995; 26: 2120-2126.
- Zhang W, Hou S-T, Stanimirovic DB. Blood-brain barrier protection in stroke: Taming tPA. In. The Blood-Brain Barrier in Health and Disease (Volume two). Ed. K. Dorovini-Zis, CRC press/Taylor & Francis Group, New York. 2015a; 226-246.
- Fujimura M, Gasche Y, Fujimura-Morita Y, Massengale J, Kawase M, Chan PK. Early appearance of activated matrix metalloproteinase-9 and blood brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999; 842: 92-100.
- Montaner J, Avarez-Sabin J, Molina C, Angeles A, Abilleira S, Arenillas J, et al. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile, and relation to neurological impairment. Stroke. 2005; 36: 1415-1420.
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 2000; 18: 7037-7042.
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J. Neurosci. 2005; 25: 6401-6408.
- Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998; 5: 1020-1030.
- Rosell A, Lo EH. Multiphasic roles of matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008; 8: 82-89.
- Wu HT, Lin J, Liu YE, Chen HF, Hsu KW, Lin SH, et al. Luteolin suppresses androgen receptor-positive triple-negative breast cancer cell proliferation and metastasis by epigenetic regulation of MMP9 expression via the AKT/mTOR signaling pathway. Phytomedicine. 2021; 81: 153437.
- Luo S, Li H, Mo Z, Lei J, Zhu L, Huang Y, et al. Connectivity map identifies luteolin as a treatment option of ischemic stroke by inhibiting MMP9 and activation of the PI3K/Akt signaling pathway. Exp Mol Med. 2019; 51: 1-11.
- Zhou ZB, Yang B, Li X, Liu H, Lei G. Lysophosphatidic Acid Promotes Expression and Activation of Matrix Metalloproteinase 9 (MMP9) in THP-1 Cells via Toll-Like Receptor 4/Nuclear Factor-κB (TLR4/NF-κB) Signaling Pathway. Med Sci Monit. 2018; 24: 4861-4868.
- Galis ZS, Sukhova GK, Libby P. Microscopic localization of active proteases by in situ zymography: detection of matrix metalloproteinase activity in vascular tissue. FASEB J. 1995; 10: 974–980.