
Research Article
Austin J Cerebrovasc Dis & Stroke. 2019; 6(1): 1081.
Elevated Lipocalin-2 Can Indicate the Vascular Inflammation in Patients with Ischemic Stroke
Rajnics P1,2*, Kellner A1, Nagy F3, Alföldi V3, CsBödőr C4, Gángó A4, Moizs M1 and Egyed M1
1Kaposi Mór Teaching Hospital Department of Hematology, Kaposvár, Hungary
2Faculty of Health Sciences, Doctoral School, University of Pécs, Pécs, Hungary
3Kaposi Mór Teaching Hospital Department of Neurology, Kaposvár, Hungary
4MTA-SE Lendulet Molecular Oncohematology Research Group, 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest
*Corresponding author: Rajnics P, Kaposi Mór Teaching Hospital, Department of Hematology, Kaposvár, Hungary
Received: May 29, 2019; Accepted: July 15, 2019; Published: July 22, 2019
Abstract
Purpose: Elevated level of Lipocalin-2 (LCN2), a new acute phase adipokine, was described after ischemic stroke. A number of researchers feel as though that LCN2 originated from the infiltrating neutrophils and other cells in brain after stroke. Others measured elevated LCN2 expression in arteriosclerotic plaque. Therefore we have investigated LCN2 relative gene expression level of blood neutrophil granulocytes in patients with ischemic stroke to assess if elevated LCN2 is the cause or consequence of ischemic stroke.
Methods: Laboratory and anamnestic data were collected, which could have a role in development of thrombo-embolic events in patients with ischemic stroke. RNA based method was used to evaluate the relative gene expression level of LCN2. We calculated Odds Ratio (OR) and Confidence Interval (CI) for the association between LCN2 and ischemic stroke.
Results: 34 samples were available for evaluation. The LCN 2 relative gene expression level was decreased in 12 cases. In this group, 91% of patients have Atrial Fibrillation (AF) at the time of hospitalisation. The mean LCN2 relative gene expression value was 64.25% (ranges: 34%-115%) in patients with AF. It was significantly lower than in patients with normal sinus rhythm (409.2%; ranges: 127%-1127%; p=0.0003). The elevated LCN2 relative gene expression level significantly (p=0.012) increases the risk of stroke (OR: 12.6) independently from other factors.
Conclusions: High LCN2 expression level seems to have strong positive predictive value on ischemic stroke, and may be useful in thrombotic risk stratification of plaque vulnerability in these patients.
Keywords: Lipocalin-2; Ischemic Stroke; Plaque Vulnerability; Inflammation
Introduction
Stroke is a sudden loss of neurological function due to ischemia or hemorrhage in the brain. It is responsible for many death and could be cause long-term disability, therefore contribute to worsening quality of life [1].
There are two main types of stroke: hemorrhagic and ischemic strokes. Hemorrhagic stroke results from rupture of cerebral blood vessels. Ischemic stroke is caused by blockage of blood flow into the brain by embolus or thrombus. The majority of strokes are ischemic (approx.87% of all), the remainder is hemorrhagic, but this latter accounts for 50% of stroke originated death [2-5].
The emboli or thrombi could be arise from the heart, mostly in the case of atrial fibrillation, or from an abrupted plaque located in the carotid vasculature. The plaque rupture occurs mostly in patiets with vulnerable plaques, but we have no exact method, which could be prognosticate the time of rupture.
Despite of the abundant theories of the mechanisms underlying the thrombotic state of ischemic strokes, there are no clear and definite answers to this question actually.
The scientific interest is focused on the neutrophil gelatinaseassociated lipocalin (lipocalin-2; LCN2), which has been implicated in the pathobiology of inflammation processes. LCN2 was described as an acute phase protein, which is mainly released from granules of activated neutrophils [6]. Matrix metalloproteinase-9 (MMP- 9), which provide forward progress the inflammatory cells within the tissues, could be bind to LCN2 and form a dimeric MMP-9/ LCN2 complex. The binding prevents the MMP-9 degradation, and prolongs the deleterious effects of MMP-9 on the arterial wall [7].
Within 24 hours after stroke, infiltrating neutrophils, macrophages, and T-cells release proinflammatory cytokines, chemokines, Reactive Oxygen Species (ROS), and Matrix Metalloproteinase (MMP). This immunreaction has detrimental effects, but may be needed for remodelling and repairing processes [8]. Some researchers found that plasma level of LCN2 is elevated at 1-3 days in patients with ischemic stroke. They speculated that the LCN2 is secreted by the infiltrating neutrophils and other cells in brain after stroke [9-11].
Eilenberg et al. showed an elevated LCN2 expression in human macrophages, smooth muscle cells and endothelial cells of endarterectomy specimens in vitro [12]. Others reported increased LCN2 levels in atherosclerosis which linked to inflammatory processes [13,14].
Our team showed recently that higher level of LCN2 expression in blood neutrophil granulocytes associated with thromboembolic event in patient with polycythemia and essential thrombocythemia [15].
We hypothesize that there is an inflammatory state in the body, which contribute to the cardiovascular events. Therefore we have examined the expression of LCN2 in blood neutrophil granulocytes in patients with ischemic stroke, to speculate whether the elevated LCN2 causes cardiovascular event or the post-stroke inflammation processes elevate this value. Currently, there are no proven medical prognostic sign or laboratory values of strokes, before the event, therefore we have examined the LCN2 expression whether it has a predictive value in the thrombotic events in patients with ischemic stroke.
Materials and Methods
Eligibility criteria
Eligibility criteria included: age 18 years or over; acute ischemic stroke diagnosed by expert neurologist and by acute CT and MRI of the brain.
Diagnostic processes
We have collected routine laboratory and anamnestic data (dyslipidemia, hypertension and diabetes mellitus) which could have a role in development of thrombo-embolic events. Electrocardiography was performed at the time of hospitalisation. Carotid ultrasonography and echocardiography was made within one week after stroke.
Ethics and study management
The study was conducted according to good clinical and laboratory practice rules and the principles of the Declaration of Helsinki. Informed written consent was obtained after the purpose, nature, and potential risks were explained to the subjects.
Measurement of LCN2 mRNA levels using quantitative real-time PCR (Q-PCR)
Total RNA was extracted from peripheral blood samples using Trizol reagent (Ambion) as recommended by the manufacturer. Two micrograms of RNA were reverse transcribed with the High-Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions. The Q-PCR analysis was performed using a Taqman probe based gene expression assay (Hs01008571_ m1, Life Technologies) according to standard protocols with GUSB (Hs99999908_m1) used as endogenous control. The displayed values were calculated by the ΔCT method and represent relative LCN2 expression values normalized to GUSB expression.
Statistical analysis
Commercially available statistical software (Social Science Statistics; (www.socscistatistics.com) was used to calculate statistics. Median (quartile) values were given to describe continuous variables, absolute numbers and percentages are used to describe categorical variables. Characteristics of patients with low expression LCN2 level was compared to characteristics of patients with high LCN2 expression level. Differences between these characteristics, Student t-test was performed in the case of normal distribution.
In a multivariate analysis, logistic regression was performed to adjust for sex, age, blood parameters, BMI, CRP, and cardiovascular risk factors (such as presence of dyslipidemia, hypertension or diabetes mellitus). Adjusted OR values (and 95% CIs) were also calculated.
Results
Patient characteristics
36 patients with ischemic stroke were enrolled to this prospective study. LCN 2 relative gene expression analysis was conducted in all cases. Measurement of two samples were missed because of technical problems. These patients were excluded from the data analysis. Data of 34 patients were assessed. The main characteristics of the patients are showed in Table 1.
![]()
Variables
Number / (%of all)
Variables
Mean values
SD
Male
19 / (55.8)
Age (years)
69..2
9.95
Female
15 / (44.2)
BMI (kg/m2)
25.62
1.99
Diabetes
11 / (32.3)
Hemoglobin (g/L)
130.4
16.4
Hypertension
25 / 73.5)
White blood cell (G/L)
9.54
2.64
Atrial Fibrillation
11 / (32.3)
Thrombocyte (G/L)
245.4
58.9
Renal insufficiency
1 / (2.9)
CRP (mg/L)
34.26
35.6
Smoking
12 / (35.3)
Cholesterine (mmol/L)
4.65
1.09
Periferal arterial disease
6 / (17.6)
Trigliceride (mmol/L)
1.98
1.17
Iscemic heart disease
8 / 23.5)
LCN2 gene expr. (%)
291
81.2
Carotid artery stenosis
22 / (64.7)
Table 1: The main characteristics of the patients. Abbrev: BMI: Body Mass Index; CRP: C-reactive protein; LCN2: Lipocalin-2.
LCN 2 relative gene expression results
The LCN 2 relative gene expression level was elevated in 22 cases and not elevated in 12 cases. In the latter group, 91% of patients (11/12) have Atrial Fibrillation (AF) on electrocardiography at the time of hospitalisation. The remaining patient did not have AF in anamnestic data. The mean lipocalin 2 relative gene expression value was 64.25% (ranges: 34%-115%) in patients with AF and it was significantly lower than in patients with normal sinus rhythm (409.2%; ranges: 127%- 1127%; p-value: 0.0003) (Figure 1). We compared characteristic variables and laboratory values of this two subgroups (Table 2).
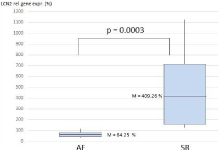
Figure 1: The Lipocalin-2 relative gene expression values in two subgroups.
The boxes show the lower and upper quartiles, the means (M) and minimum
and maximum values (lines). AF: patients with atrial fibrillation. SR: patients
with synus rhythm.
![]()
Variables
Atrial fibrillation
Normal synus rhythm
P-value
sex (M/F; %/%)
4/7 (36%/64%)
13/10 (56% / 44%)
0.804
age median (range) year
71,4 (48-88)
67.7 (60-89)
0.79
LCN2 rel.expr. median (range)
64.25 (34-115)
409.2 (127-1127)
0.0003
WBC (mean G/L; range)
7.8 (5-24)
8.4 (4-22)
0.07
Hb (mean g/L; range)
128 (102-166)
134 (105-170)
0.085
PLT (mean G/L; range)
235 (205-371)
248 (135-352)
0.666
BMI (mean kg/m2; range)
25.1 (21-30)
26.3 (20-38)
0.09
CRP (mean mg/L; range)
24.12 (0.15-32)
36.4 (0.2-89)
0.24
Dyslipidaemia (n, %)
4 (36%)
11 (47.8%)
0.68
Hypertension (n, %)
7 (63%)
13 (56%)
0.47
Diabetes mellitus (n, %)
3 (27%)
6 (26%)
0.89
Table 2: Statistical comparison of characteristic variables and laboratory values of patients with atrial fibrillation and normal synus rhythm.
There was no significant difference between two subgroups, except in the fact that in lower LCN2 subgroup the patients have AF at the time of stroke. In this group 5 patient have carotid artery plaque, which caused significant stenosis.
We have assessed a multivariate analysis that includes LCN2 and other clinical and laboratory variables previously shown to impact stroke risk (Table 3). According to our results the elevated LCN2 relative gene expression level significantly (p=0.012) increases the risk of stroke (OR: 12.6) independently from other factors.
![]()
95% CI for OR
Variables
B
S.E.
Wald
df
p-value
OR
Lower
Upper
Age
0.025
0.024
0.666
1
0.51
1.031
0.941
1.091
Sex
0.019
0.63
0.002
1
0.892
1.036
0.234
3.442
Lcn2
2.51
1.177
5.812
1
0.012
12.671
1.598
84.79
WBC
0.018
0.802
0.013
1
0.894
1.122
0.199
5.469
PLT
0.019
0.022
1.039
1
0.345
1.022
0.968
1.012
BMI
-0.977
0.572
3.246
1
0.066
0.353
0.141
1.34
CRP
0.136
0.067
1.671
1
0.154
1.133
0.889
1.356
Dyslipidemia
-0.044
0.657
0.004
1
0.905
0.961
0.284
3.32
Hypertension
0.945
0.678
2.032
1
0.163
2.537
0.812
9.534
Diabetes mellitus
0.387
0.677
0.175
1
0.781
1.411
0.432
5.745
Table 3: Multivariate analysis of clinical and laboratory variables previously shown to impact stroke risk and LCN2. Abbrev.: LCN2: Lipocalin-2; WBC: white blood cell; PLT: Platelet; BMI: Body Mass Index; CRP: C-reactive protei.
Discussion
Stroke is a leading cause of adult disability in the western countries. Ischemic stroke developed after blockage of blood flow into the brain by thrombus or embolus. Ischemia initiates cerebral infarction during ischemic stroke, but reperfusion after recanalization may promote secondary injury and worsen neurological outcomes [16,17]. Multiple pathophysiological pathways have been identified in the development of vascular inflammation and thrombosis, which contribute to aggressive atherosclerosis [18,19].
Lipocalins are multifunctional proteins recognized as carriers of small hydrophobic molecules [20]. A common feature is their β-sheet tertiary structure, consisting of eight antiparallel strands, which form a calyx with a hydrophobic cavity [21]. LCN 2 is originated from neutrophils, which contain a variety of granules (secretory vesicles, gelatinase granules, azurophil granules) [22,23].
Animal studies showed that LCN 2 was elevated after transient middle cerebral artery occlusion (tMCAO) in rodents [24-26]. LCN2 appears in mouse sera as early as one hour, peaks at 23 hours, and diminishes by 48 to 72 hours after tMCAO. [27] Xing et al. descibed earlier that 3 days after an ischemic stroke in rats and humans, LCN2 is expressed in injured neurons. They hypotesized that this is a “help me signal” to condition microglia and astrocytes for recovery [26].
Liu and coworkers induced endothelial disfunction by lipocalin 2 treatment in animals. The oxidative stress and endogene nitrogeneoxide synthase (eNOS) uncoupling were accountable for this effect [28].
Eilenberg et al. showed that LCN2 is expressed by macrophages, smooth muscle cells and endothelial cells in human carotid plaques [12,29]. There is an alternative possible mechanism by which LCN2 affects the arterial wall. LCN2 forms a dimeric MMP-9/NGAL complex, by prevents the MMP-9 degradation, so it can prolong the deleterious effects of MMP-9 on the arterial wall [7].
We have hypothesized that elevated LCN 2 level is not a consequence of ischemic stroke, but a cause of plaque rupture in a destroyed cardiovascular system. We have examined the LCN 2 relative gene expression level in blood granulocytes in patients with ischemic stroke. It was found that higher relative expression of this gene was measured in majority of patients with ischemic stroke. The values were lower in patients with ischemic stroke with atrial fibrillation.
Our measurement on gene expression level was made from blood neutrophil granulocytes and support the fact that there is an inflammatory environment in the blood vessels without leukocytosis and elevated CRP around the stroke event. This inflammatory potential was not observed in majority of patients, who have AF at the time of stroke. Their stroke developed in other way, perhaps by embolisation from the heart. However we could not detect any intracavital thrombus in heart by echocardiography.
Wirchow’s triad have role in thrombus formation in the left atrium. Many researchers described the blood stasis, endothelial dysfunction and clotting activation in AF. It is evident that reduced flow velocity and impaired contractility of left atrial wall cause blood stasis [30]. Higher level of von Willebrand Factor (vWf) and E-selectin was measured in blood of patients with AF, revealed to endothelial dysfunction [31,32]. Finally, AF may cause a hyper-coagulation state, which was measured by increase of plasma levels of D-dimer and fibrinogen and provide the evidence of clotting activation in this patients [33,34].
As a carrier, LCN 2 may affect polyamine homeostasis within the vasculature. Polyamination of molecules in endothelial cells, such as RhoA, an element of a kinase pathway, plays an important role in endothelial integrity and function. Because polyamines and NO are originated from the common precursor, arginine, changes
of polyamine metabolites may influence the eNOS/NO metabolic pathway and leading to endothelial dysfunction and vascular inflammation [35]. Now it is well known that ROS are affecting endothel cells impaired in their function, therefore disrupt the balance between antithrombotic and thrombotic factors.
Conclusions
According to our pilot study the elevated LCN2 gene expression in periferal blood granulocytes confirm a proinflammatoric state throughout the body, which contribute to the disruption of vulnerable plaque and consequent trombosis and microembolisation.
Additional studies needed to evaluate whether LCN2 may serve as potential blood biomarker of plaque vulnerability in patients with cardiovascular diseases. Respectively to judge if it may be a reliable blood biomarker in early detection of stroke.
References
- Lees KR. Management of acute stroke. Lancet Neurol. 2002; 1: 41-50.
- van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet. 2007; 369: 306-318.
- Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009; 373: 1632-1644.
- Rincon F, Mayer SA. Intracerebral hemorrhage: Clinical overview and pathophysiologic concepts. Transl Stroke Res. 2012; 3: 10-24.
- Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012; 11: 720-731.
- Liu Q, Nilsen-Hamilton M. Identification of a new acute phase protein. J. Biol Chem. 1995; 270: 22565-22570.
- Yan L, Borregaard N, Kjeldsen L, Moses MA. The high molecular weight urinary Matrix Metalloproteinase (MMP) activity is a complex of gelatinase B/ MMP-9 and Neutrophil Gelatinase-Associated Lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J Biol Chem. 2001; 276: 37258-37265.
- Eltzschig HK, Eckle T. Ischemia and reperfusion from mechanism to translation. Nat Med. 2011; 17: 1391-1401.
- Elneihoum AM, Falke P, Axelsson L, Lundberg E, Lindgarde F, Ohlsson K. Leukocyte activation detected by increased plasma levels of inflammatory mediators in patients with ischemic cerebrovascular diseases. Stroke. 1996; 27: 1734-1738.
- Anwaar I, Gottsater A, Ohlsson K, Mattiasson I, Lindgarde F. Increasing levels of leukocyte-derived inflammatory mediators in plasma and camp in platelets during follow-up after acute cerebral ischemia. Cerebrovasc Dis. 1998; 8: 310-317.
- Falke P, Elneihoum AM, Ohlsson K. Leukocyte activation: Relation to cardiovascular mortality after cerebrovascular ischemia. Cerebrovasc Dis. 2000; 10: 97-101.
- Eilenberg W, Stojkovic S, Piechota-Polanczyk A, Kaun C, Rauscher S, Groger M. Neutrophil Gelatinase-Associated Lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro-inflammatory state in vitro. Eur J Vasc Endovasc Surg. 2016; 51: 623-631.
- Leclercq A, Houard X, Philippe M, Ollivier V, Sebbag U, Meilhac O, et al. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol. 2007; 82: 1420-1429.
- te Boekhorst BC, Bovens SM, Hellings WE, van der Kraak PH, van de Kolk KW, Vink A, et al. Molecular MRI of murine atherosclerotic plaque targeting NGAL: a protein associated with unstable human plaque characteristics. Cardiovasc Res. 2011; 89: 680-688.
- Rajnics P, Kellner A, Karadi E, Moizs M, Bödör Cs, Király PA, et al. Increased Lipocalin 2 level may have important role in thrombotic events in patients with polycythemia vera and essential thrombocythemia. Leukemia Research. 2016; 48:101-106.
- Iadecola C, Anrathe J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011; 17: 796-808.
- Chou WH, Messing RO. Protein kinase c isozymes in stroke.Trends Cardiovasc Med. 2005; 15: 47-51.
- Pircher A, Treps L, Bodrug N, Carmeliet P. Endothelial cell metabolism: a novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis. 2016; 253: 247-257.
- Taleb S. Inflammation in atherosclerosis. Arch Cardiovasc Dis. 2016; 109: 708-715.
- Flower DR. The lipocalin protein family: structure and function Biochem. J. 1996: 1-14.
- Flower DR. Structural relationship of streptavidin to the calycin protein superfamily FEBS Lett. 1993; 333: 99-102.
- Borregaard N. Development of neutrophil granule diversity. Ann N Y Acad Sci. 1997; 832: 62-68.
- Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005; 123: 1293-1300.
- Chiang T, Messing RO, Chou WH. Mouse model of middle cerebral artery occlusion. J Vis Exp. 2011; 13.
- Jin M, Kim JH, Jang E. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2014; 34: 1306-1314.
- Xing C, Wang X, Cheng C. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke. 2014; 45: 2085-2092.
- Wang G, Weng YC, Han X, Whaley JD, McCrae KR, Chou WH. Lipocalin-2 released in response to cerebral ischaemia mediates reperfusion injury in mice. J Cell Mol Med. 2015.
- Liu JT, Song E, Xu A, Berger T, Mak TW, Tse HF, et al. Lipocalin-2 deficiency prevents endothelial dysfunction associated with dietary obesity: role of cytochrome P450 2C inhibition. Br J Pharmacol. 2012; 165: 520-531.
- Eilenberg W, Stojkovic S, Kaider A, Kozakowski N, Domenig CM, Burghuber C. NGAL and MMP-9/NGAL as biomarkers of plaque vulnerability and targets of statins in patients with carotid atherosclerosis. Clin Chem Lab Med. 2017.
- Watson T, Shantsila E, Lip GY. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet. 2009; 373: 155-166.
- Nightingale T, Cutler D. The secretion of von willebrand factor from endothelial cells; an increasingly complicated story. Journal of thrombosis and haemostasis: JTH. 2013; 11: 192-201.
- Krishnamoorthy S, Khoo CW, Lim HS, Lane DA, Pignatelli P, Basili S, et al. Prognostic role of plasma von willebrand factor and soluble e-selectin levels for future cardiovascular events in a ‘real-world’ community cohort of patients with atrial fibrillation. European journal of clinical investigation. 2013; 43: 1032-1038.
- Mitusch R, Siemens HJ, Garbe M, Wagner T, Sheikhzadeh A, Diederich KW. Detection of a hypercoagulable state in nonvalvular atrial fibrillation and the effect of anticoagulant therapy. Thrombosis and haemostasis. 1996; 75: 219- 223.
- Heppell RM, Berkin KE, McLenachan JM, Davies JA. Haemostatic and haemodynamic abnormalities associated with left atrial thrombosis in nonrheumatic atrial fibrillation. Heart. 1997; 77: 407-411.
- Lee DL, Webb RC, Jin L. Hypertension and RhoA/Rho-kinase signaling in the vasculature: highlights from the recent literature. Hypertension. 2004; 44: 796-799.