
Research Article
Austin Chem Eng. 2020; 7(2): 1077.
5'-Dimethoxytrityl-Protected Double-Stranded DNA: Synthesis and Competence to Blunt-Ended Ligation and 5', 3'-Exonuclease Activity
Shchur VV, Burankova YP, Dzichenka YV, Usanov SA and Yantsevich AV*
Institute of Bioorganic Chemistry, National Academy of Sciences, Belarus
*Corresponding author: Yantsevich Aleksei V, Institute of Bioorganic Chemistry, National Academy of Sciences, Belarus
Received: June 15, 2020; Accepted: July 03, 2020; Published: July 10, 2020
Abstract
The functionalization of 5'-OH group in nucleic acids is of significant value for molecular biology. For example, asymmetric phosphorylation of sense and antisense strands of DNA found application during the generation of gene-size ssDNA, that isimportant nowadays for precise editing of genomes by CRISPR/Cas9 methodology. In the current work we discovered that acid-labile 4,4'-Dimethoxytrityl Protecting Group (DMT) of oligonucleotides is stable under PCR conditions and does not interfere with activity of DNA polymerases. So application of 5'-DMT-protected oligonucleotides could allow producing both symmetric and asymmetric 5'-DMT-blocked DNA fragments. We demonstrated that the presence of thiol compounds (mercaptoethanol and dithiothreitol) in PCR mixture is undesirable for the stability of DMT-group. DMT-oligonucleotides can be successfully used during polymerase chain assembly of synthetic genes. We tested 5'-DMT dsDNA in blunt-end DNA ligation reaction by T4 DNA ligase and found that it could be effectively ligated with 5'-phosphorylated DNA fragments, namely linearized plasmid vector. We alsodemonstrated that5'-DMT modification of dsDNA does affect activity of T5 5',3'-exonuclease towards both ssDNA and dsDNA. Further search of the exonucleases, sensitive to 5'-DMT-modification or ways to separate 5'-DMT-ssDNA and 5'-OH-ssDNA could allow finding application of 5'-DMT-modified oligo- and polynucleotides.
Keywords: Oligonucleotide; DMT,4,4'-dimethoxytritylprotection group,Long ssDNA; Synthetic gene; Cloning.
Introduction
Functionalization of a 5'-hydroxyl group of nucleic acids founds many applications in DNA technology [1], because it allows following and detecting labeled molecules and to control specificity of the enzymes.
For example, stability, uptake and immunogenicity of therapeutic siRNA and mRNA could be improved by structural modification of the 5'-end [2]. Phosphorylation by T4 polynucleotidekinase is used for tagging of oligonucleotides before sequencing, PCR and ligation [3]. In the latter case,selective ligation of phosphorylated DNA fragments into the dephosphorylated vector happens (Figure 1) [4].
Asymmetric labeling of 5'-end of DNA is of particular importance for the generation of “gene-size” single-stranded DNA (ssDNA) [5]. Currently, ssDNA is used as a donor template for knocking experiments requiring DNA fragments with length more than 500 bases [6]. ssDNA repair templates have recently been shown to have advantages over dsDNA. Among them are a very low tendency to random genome integration [7], low cytotoxic response and absence of expression from nonintegrated templates, that facilitates correct identification of genome edited clones. At the same time, the usefulness of long ssDNA templates is limited due to the difficulty and cost of producing them.
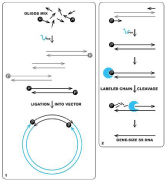
Figure 1: Exploring 5'-OH modifications of nucleic acids in molecular biology. 1. Selective ligation of full-length sequence from a mixture of possible synthetic gene assembly products. Only flanking oligonucleotides are phosphorylated, vector does not have 5′-phosphate and could not be self-ligated; 2. The general way to obtain gene-size ssDNA by asymmetrically labeled oligonucleotides. Currently used phosphorylating and splitting kit used to destroy phosphorylated DNA strand.
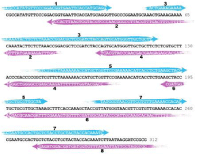
Figure 2: Design of 312 bp DNA sequencewas obtained from eight oligonucleotides by PCA. Design was generated by DNAWorks software11. Parameters for DNAWorks: Oligonucleotide Size- 66 nt, Annealing Temp-65 °C.
One solution to generate ssDNA templates is to convert a dsDNA PCR product into ssDNA by selectively digesting either the sense orthe antisense strand.Selective enzymatic degradation of one strand of DNA could be achieved by labeling of 5'-OH (for example, by phosphorylation), as shown on Figure 1-2. Such an idea was realized using a strandase enzyme-based approach from Takara Biosciences. Purely enzymatic synthesis to generate ssDNA greater than 15 kb using asymmetric PCR was reported [5].
So, search of alternative ways for 5'-OH modification (symmetric or asymmetric) of DNA and investigation of functional competence of such modifications to enzymatic reactions is of importance for the generation of “gene-size” ssDNA, demand for which will increase in the next years.
[1] Selective ligation of full-length sequence from a mixture of possible synthetic gene assembly products. Only flanking oligonucleotides are phosphorylated, vector does not have 5′-phosphate and could not be self-ligated [2]. The general way to obtain gene-size ssDNA by asymmetrically labeled oligonucleotides. Currently used phosphorylating and splitting kit used to destroy phosphorylated DNA strand.
Owing to the low reactivity of the 5'-hydroxyl, its modification with the required functional group is not easily achieved without side reactions at the nitrogen base level. So in order to functionalize5'-end of dsDNA such modification should be introduced on the stage of oligonucleotide synthesis1. Subsequent PCR will give 5'-functionalized dsDNA fragment.
During oligonucleotide synthesis orthogonal protection systems must be used, most of which are based on acid-labile 4,4'-dimethoxytrityl (DMT) protective group. Since its introduction as acid-labile protecting group for deoxyribonucleotide chemistry, DMT becomes standard for the protection of the 5'-OH group8.Usually deblocking is realized by 2-3% trichloroacetic acid solution in toluene ordichloromethane. After last base coupling, DMT could be removed (DMT-OFF synthesis) or left (DMT-ON synthesis) on the oligonucleotide.
Hydrophobic properties of DMT allows blocked oligonucleotide to be retained on reverse-phase sorbents in the presence of trimethylamine as an ion-pairing reagent. So fast and effective procedures for oligonucleotide purification based on reverse-phase solid-phase extraction cartridgeswere developed. During purification, DMT is removed by washing with 2%trifluoroacetic acid.Earlier we demonstrated the ability of ZipTip microextractors to purify and dose oligonucleotide pools for subsequent gene assembly [9].
The application of DMT-oligonucleotides in PCR and during gene assembly was not reported earlier. Thus effectiveness of DMT-oligonucleotides for PCR amplification or for gene assembly remains unknown as well as the fate of the protective group after PCR.
Further investigations of influence of bulky, lipophilic DMT-group on activity and specificity of DNA-enzymes, especially 5',3'-exonucleases, will allow outlining the prospects for practical application of 5'-DMT dsDNA.
Materials and Methods
Materials
5′-DMT-dABz-, 5′-DMT-dT-, 5′-DMT-dCAc-, and 5′-DMT-dGiBu-CPG with a pore size of 1000 Å and loading capacity of 42–45 μmol/g were from BioAutomation (Irving, TX). DMT-dABz, DMT-dGiBu, DMT-dCBz, and DMT-dT phosphoramidites; dichloromethane; acetic anhydride; lutidine; 1-methylimidazole; trifluoroacetic acid; and methanol were from Sigma-Aldrich (St. Louis, MO). HPLC-grade acetonitrile with low water content (0.002%) was from Fisher Chemical (Waltham, MA). 5-(Ethylthio)-1H-tetrazole (Berlin, Germany). 10?DreamTaq buffer (for DreamTaq Polymerase) and dNTP mix were from Thermo Scientific (Waltham, MA). Q5 High-Fidelity DNA polymerase, 100bp and 1kb Plus DNA ladder werepurchased from New England Biolabs (Ipswich, MA). 32% ammonia solution was from Roth (Karlsruhe, Germany). Triethylamine was from Alpha Aesar (Kandel, Germany).
General methods
A260 and electronic spectra for oligonucleotide solutions were recorded with Nano Drop UV-Vis spectrophotometer from Thermo Scientific (Waltham, MA).
PCR based gene assembly and oligonucleotide annealing were performed in thermocycler DNA Engine (Bio Rad Laboratories, Hercules, CA).
Agarose gelelectrophoreses were performed at 105V. Visualization of gel electrophoresis was made using Azure Biosystems c280 geldocumentation system (Azure Biosystems, Dublin, CA).
Oligonucleotide preparation
Common oligonucleotides were synthesized, deprotected and purificated as described in [10]. DMT-protected oligonucleotides were purified by the same procedure, except the detritylation step with trifluoroacetic acid: after washing with 11% ACN in 0.1 M TEAAc (pH 8.0) buffer oligonucleotides were eluted in 20% ACN and vacuum evaporated. In order to minimize detritylation, 50µl of concentrated aqueous ammonia was added to eluate before evaporation.
HPLC-MS analysis
Analytical HPLC analysis with a Diode Array Detector (DAD) and MS detector was performed on an Agilent 1290 HPLC system according to the application note “Characterization of Synthetic Oligonucleotides Using Agilent LC/MS Systems” from Agilent.
![]()
?
Oligonucleotide
Length
Position in sequence
1
CGCCATATGTTCCCGGACGGTGAATTCACCATGCAGG
37
Jan-37
2
GGTTTAGAGAAGTATTTGTTTTCTTTCAGTTTGCATTCCGGGCAACCCTGCATGGTGAATTCACCG
66
18-83
3
ACTGAAAGAAAACAAATACTTCTCTAAACCGGACGCTCCGATCTACCAGTGCATGGGTTGCTGCTT
66
54-119
4
CCAGCATGGTTTTTTTAGAACGAGCCGGGGTCGGGTAAGCACGAGAGAAGCAGCAACCCATGCACT
66
101-166
5
CTCGTTCTAAAAAAACCATGCTGGTTCCGAAAAACATCACCTCTGAAGCTACCTGCTGCGTTGCTA
66
143-208
6
GTTTTCAACACGAACGTTACCCATAACGGTAGCTTTGGTGAAAGCTTTAGCAACGCAGCAGGTAGC
66
190-255
7
TATGGGTAACGTTCGTGTTGAAAACCACACCGAATGCCACTGCTCTACCTGCTACTACCACAAATC
66
231-296
8
CGCGGATCCTTATTAAGATTTGTGGTAGTAGCAGGTAGAG
40
273-312
Table 1: List of oligonucleotidesused for enzymatic assembly of synthetic DNA fragment.
![]()
Name
Sequence(5'�3')
DNA65_1 sense
ATCTTCTCAATTGACCTTGCGAATTCCCCCCCATGGTAGCTCGAAGCTTATCACCTGCTTTCGAT
DNA65_2 antisense
ATCGAAAGCAGGTGATAAGCTTCGAGCTACCATGGGGGGGAATTCGCAAGGTCAATTGAGAAGAT
Solvent
Content of 5'-DMT-dT15
1 min
5 min
15 min
Water
44,5%
7,43%
0%
1? Q5 DNApolymerase buffer
-
88,4%
71,5%
1?DreamTaqDNApolymerase buffer
-
93,5%
83,69%
25 PCR cycles in 1?DreamTaq DNA polymerase buffer
79,5%
Table 2: Content of 5′-DMT-dT15 oligonucleotide in different solutions after heating at 98°C.
Phase A was 200mM HFIP in 0.1M TEAAc buffer (pH 7.7), phase B wasmethanol. Solvent rate was 0.2 ml/min. Gradient of phase B from 5 to 35% during 30 min was used for separation. The detection wavelength of DAD detector was set to 260 nm. High-resolution mass spectra were registered with a Q-TOF 6550 high-resolution mass spectrometer (Agilent) in the ESI-mode. Mass spectra of oligonucleotides were deconvoluted by Bio Confirm module of Mass Hunter software.
Design and assembly of synthetic DNA fragment
As a model synthetic gene we used 312bp sequence:
1 CGCCATATGTTCC CGGACGGTGAATTCACCAT GCAGGGTTGCCCGGAA TGCAAACTGAAA
61 GAAAACAAAT ACTTCTCTAAA CCGGACGCTC CGATCTACCAG TGCATGGGTTGCTGCTTC
121 TCTCGTGCTTACC CGACCCCGGCTC GTTCTAAAAAAACC ATGCTGGTTCCGAAAAACATC
181 ACCTCTGAAGCTAC CTGCTGCGTT GCTAAAGCTTT CACCAAAGCTA CCGTTATGGGTAAC
241 GTTCGTGTTGAAA ACCACACCGAATGCC ACTGCTCTACCT GCTACTACCA CAAATCTTAA
301 TAAGGATCCGCG
Generation of short DMT-protected dsDNA fragment by direct oligonucleotide annealing
In order to obtain short DMT-blocked dsDNA fragment (65bp), we synthesized two complementary oligonucleotides (Table 2). Oligonucleotides were designed to double EcoRV restriction site (bold type in table 2) in order to allow easy control of fragment insertion.
Oligonucleotides were ammonia deprotected and purified in DMT-ON mode as stated above. Purified oligonucleotide solutions were aliquoted and vacuum evaporated for storage. 50µl of aqueous ammonia were added to the oligonucleotide solution before evaporation in order to prevent the loss of DMT-group.
For dsDNA preparation, we used solutions of DMT-oligonucleotides in deionized water.
For annealing of oligonucleotides we used the following program: 3 min at 95°C then cooling to 20°C at a rate of 0.1°C per second.
For DMT-OFF dsDNA 15µl of water, 3.75µl of each DMT-oligonucleotide solution were mixed in PCR tube. PCR tube was placed in thermocycler and heated and cooled according to the annealing program described above. At this stage DMT-oligos loss DMT group. Then 2.5µl of 10?DreamTaqbuffer was added, mixed and repeatedly annealed.
In order to get 5′-DMT dsDNA 15µl of water, 3.75µl of each DMT-oligonucleotide solution and 2.5µl of 10?DreamTaqbuffer were mixed in PCR tube. The annealing program was repeated twice in order to ensure equal conditions with the previously described procedure for DMT-OFF dsDNA generation.
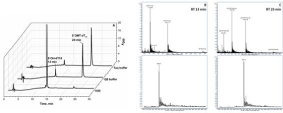
Figure 3: Heat-activated detritylation of DMT-dT15 oligonucleotides in water and Q5 DNA polymerase and DreamTaq DNA polymerase buffers as observed by HPLC with detection at 260 nm (A). Mass-spectra ofpeaks eluted near 13 and 23 min presented at B and C and correspond to 5′-OH-dT15 and 5′-DMT-dT15 oligonucleotides.
Blunt-ended ligation to plasmid vector
For blunt-ended ligation we used commercially available Clone JET PCR Cloning Kit from Thermo Scientific. Ligation was performed according to supplier recommendation. The molar ratio of dsDNA fragment was 3:1. Ligation was controlled by electrophoresis in 1% agarose gel and by transformation of competent E. coli cells (strain DH5α). For ligation we used short (65bp) synthetic dsDNA obtained by oligonucleotide annealing and amplification product of a synthetic gene (312bp) obtained by polymerase chain assembly as described above.
In order to easily control insert for amplification of synthetic gene, we used primers, designed to double EcoRV site (bold type):
5'-ATCCGCCATATGT TCCCGGACGGT-3' and 5'ATCCGCGGATCCTTATT AAGATTTGTGGTA-3'.
Sensitivity to 5',3'-exonuclease activity
In order to compare the sensitivity of 5'-OH-dsDNA, 5'-DMT dsDNA and 5'-DMT-oligonucleotides to T5 5', 3'-exonuclease activity, we mixed 41.7µl H2O, 5µl 10? buffer N4, 3µl of DNA solution (15pM for each chain) and 0.3µl T55',3'-exonuclease (10 U/µl) in PCR tube and incubate 30 min at 37°C.
For the analysis, we used electrophoresis in 3% agarose gel.
Results And Discussion
Heat-activated detritylation of oligonucleotides
In order to simplify stability investigation of DMT protecting group, we used 5′-DMT-dT15 oligonucleotide as a model.
HPLC analysis demonstrated that in deionized water more than 90% of oligonucleotide converts to unprotected form during short 5 min heating at 99°C (Figure 3, Table 2).
Commercially available PCR buffers tested by us demonstrated a significant effect in stabilizing DMT-oligonucleotides.
Even 15 min of heating at 98°C in 1?DreamTaqpolymerase buffer allows saving more than 83% DMT-protected dT15. Buffer for Q5 polymerase showed worse results-about 71% of DMT-protected dT15.
Analysis of PCR buffer composition (table 3) demonstrated, that tested buffers differ in pH values: Q5 buffer (pH 9.3) has a higher pH than Dream Taqbuffer (pH 8.3).
![]()
DNA polymerase
pH
1?buffer composition
Supplier
Taq
pH 8.3 (25°C)
10 mM Tris-HCl
Sigma-Aldrich
50 mM KCl
1.5 mM MgCl2
0.01% gelatin
Pfu
pH 8.8 (25°C)
20 mM Tris-HCl
Promega
10 mM KCl
10 mM (NH4)2SO4
2 mM MgSO4
1,0% TritonX-100
1mg/ml nuclease-free BSA
Pwo
pH 8.85 (20°C)
10 mM Tris-HCl
Roch
25 mM KCl
5 mM (NH4)2SO4
2 mMMgSO4
Tth
pH 8.9 (25°C)
10 mM Tris-HCl
Sigma-Aldrich
1.5 mM MgCl2
0.1 M KCl
500 μg/ml BSA
0.5% Tween 20
Q5
pH 9.3 (25°C)
25 mM TAPS-HCl
NEB
50 mM KCl,
2 mM MgCl2
1 mM β-mercaptoethanol
Phusion
pH 9.3 (25°C)
25 mM TAPS-HCl
NEB
50mM KCl
2 mM MgCl2
1 mM β-mercaptoethanol
Table 3: Content of commercially available PCR buffers (supplier information).
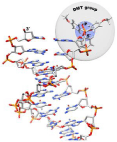
Figure 4: Superimposed structures of 5′-phosphorylated and 5′-DMT-modified dsDNA. General volumes occupied by phosphate and DMT are shown as blue and gray spheres respectively. Molecular graphics and analyses performed with UCSF Chimera.
Given the nature of the DMT group, elevated pH should further stabilize DMT-oligonucleotide, but in the real experiment, we obtained the opposite result. This contradictory result could be explained by the presence of β-mercaptoethanol in the Q5 DNA polymerase buffer.
Unwanted retritylation, that can occur through the reversal of the equilibrium generated by nonaqueous detritylation, between the colored cationic species and the DMT-oligonucleotide was reported earlier [12]. Allowing such equilibrium under heating, we could propose that such a strong nucleophile as a sulfur atom of mercaptoethanol could be a good acceptor of trityl-cation and could shift the balance to the side of the 5′-OH-oligonucleotide formation.
Synthetic DNA assembly from 5'-DMT-oligonucleotides
Superimposed structures of 5′-phosphorylated and 5′-DMT-modified dsDNA (Figure 4) suggest that bulky, hydrophobic DMT group could change the specificity of enzymes that recognize DNA endand catalyze DNA end modification reactions. So in order to check some promising reactions, we proposed three ways of generation of 5′-DMT-modified dsDNA: annealing of 5′-DMT-oligonucleotides for short sequences (<100bp), polymerase chain assembly by Stemmer approach [13] from 5′-DMT-oligonucleotides for middle size sequences (<500 bp), and amplification of target sequence with 5′-DMT-primers for DNA with length more than 300bp [14].
The result of 312bp synthetic DNA fragmentassembly from 8 oligonucleotides is shown in Figure 5. PCA from all DMT-oligonucleotides, common oligonucleotides, or from inner DMT-oligonucleotides and outer oligonucleotides showed no differences as observed in 1% agarose gel electrophoresis.
Generation of short DMT-protected dsDNA fragment agarose electrophoresis in 4% gel showed that 5′-DMT dsDNA had lower electrophoretic mobility as compared to natural DNA (Figure 6).
Blunt-ended ligation of 5'-DMT dsDNA
To test the ability of T4DNA ligase to perform blunt-ended ligation of 5′-DMT dsDNA fragments to vector we used 5′-DMT dsDNA, obtained in different ways:
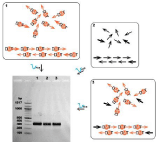
Figure 5: Agarose gel electrophoresis (1%) of “one-pot” polymerase chain assembly of 312bp synthetic DNA fragment from 8 oligonucleotides. The concentrations of oligonucleotides in assembly mixture were 20nM for oligonucleotides 2-7 and 150nM for flanking oligonucleotides 1and 8. 1. Gene assembly from DMT-oligonucleotides; 2. Assembly from deblocked oligonucleotides; 3. Assembly from inner DMT-oligonucleotides and flankingde blocked oligonucleotides.
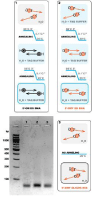
Figure 6: Agarose gel electrophoresis (4%) of dsDNA fragment obtained by oligonucleotide annealing. 1. dsDNA; 2. 5′-DMT dsDNA; 3. Oligonucleotide mixture without annealing.
- Short fragment of 65bp obtained by annealing of oligonucleotides,
- Fragment of 312bp obtained by PCR amplification of synthetic gene using DMT-blocked primers.
Both fragments were designed to double site of EcoRV restriction enzyme, so the correctness of the insertion could be easily controlled by restriction analysis.
Analysis of ligation mixture by agarose gel electrophoresis (Figure 7) allowed us to confirm blunt-ended ligation of pJET1.2/blunt with 5′-OHdsDNA as well as 5′-DMT dsDNA.
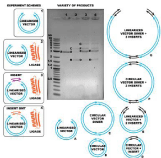
Figure 7: Ligation of test dsDNA fragment (65bp) into pJET1.2/blunt cloning vector ; 1. pJET1.2/blunt; 2. pJET1.2 +T4 DNA ligase; 3. pJET1.2 +5′OH-dsDNA +T4 DNA ligase; 4. pJET1.2 +5′-DMT dsDNA +T4 DNA ligase.
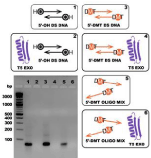
Figure 8: Exposure of 5'-OHdsDNA, 5'-DMT dsDNA and 5'-DMToligonucleotide mix to T5 5', 3'-exonuclease. 1. 5'-OHdsDNA; 2. 5'-OHdsDNA+T5 exonuclease; 3. 5'-DMT dsDNA; 4. 5'-DMT dsDNA+T5exonuclease; 5. 5'-DMT-oligonucleotide mix; 6. 5'-DMT-oligonucleotide mix + T5 exonuclease.
E. coli transformation by ligation mixture
Transformation of competent E. coli cells by ligation mixtures showed positive results for 5′-DMT dsDNA and 5′-OH-dsDNA on both cases of synthetic gene and annealed oligonucleotides. Subsequent isolation of the corresponding plasmid from night culture and restriction analysis by EcoRV enzyme confirmed insert correctness (data not shown).
Moreover, we observed higher transformation efficiency for plasmids with ligated 5′-DMT dsDNA. But the explanation of the fact lies beyond this article’s scope.
Exposure to 5',3'-exonuclease activity
T5 exonuclease degrades DNA in the 5' to 3' direction and is able to initiate nucleotide removal from the 5'-termini or at gaps and nicks of linear or circular dsDNA. As follows from the Figure 7, T5exonuclease did not show specificity to structure of 5'-termini: 5'-DMT dsDNA is degraded by T5 exonuclease as well as 5'-OHdsDNA and 5'-DMToligonucleotides (Figure 8).
Conclusion
In the current study, we confirmed that the DMT group remains on oligonucleotides in PCR conditions. The presence of thiol-compounds in PCR buffer should be avoided to maximize yield of 5'-DMT dsDNA.
We demonstrated that DMT-oligonucleotides could be efficiently used for gene assembly. So usage of DMT-oligonucleotides in PCR could be a way to generate symmetrically or asymmetrically 5'-DMT-protected dsDNA. We showed that presence of DMT has little effect on DNA electrophoretic mobility in agarose gel, which could be observed only for short fragments.
We assumed that the bulky DMTprotecting group could be a modifier of activity for enzymes that contact 5'-function of dsDNA, which would be useful for selective ligation or generation of ssDNA.
In order to test functional competence of 5'-DMT dsDNA to the enzymatic reaction, we examined dsDNA in blunt-ended ligation reaction catalyzed by T4 DNA ligase and in specific hydrolysis catalyzed by T5exonuclease. Despite our assumptions, we found that the presence of 5'-DMTprotection does not influence T4 DNA ligase activity in blunt-ended reaction and hydrolysis by T5exonuclease. Moreover, ligation products derived from 5'-DMT dsDNA unexpectedly demonstrated higher transformation efficiency as compared to common PCRproduct ligation, but an explanation of the fact is beyond the current work.
Further work on the investigation of biological properties of 5'-DMT dsDNA, including testing of the DNA-enzymes, is required to test its application in molecular biology.
Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.The authors acknowledge A. Karpenko and A. Zholnerovich for the excellent technical assistance.
This work was supported by National Academy of Sciences of Belarus (Grant No: 20191477 and Grant No: 20162001).
References
- Miller GP, Kool ET. Versatile 5'-functionalization of oligonucleotides on solid support: amines, azides, thiols, and thioethers via phosphorus chemistry. The Journal of organic chemistry. 2004; 69: 2404-10.
- Wang LK, Lima CD, Shuman S. Structure and mechanism of T4 polynucleotide kinase: an RNA repair enzyme. The EMBO journal. 2002; 21: 3873-80.
- Hoffman LM, Jendrisak J. Heat-labile phosphatase simplifies the preparation of dephosphorylated vector DNA. Gene. 1990; 88: 97-9.
- Veneziano R, Shepherd TR, Ratanalert S, Bellou L, Tao C, Bathe M. Synthesis of gene-length single-stranded DNA. Scientific reports. 2018; 8: 6548.
-
(c) Davis L, Maizels N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111: 924-32.
- Schaller H, Weimann G, Lerch B, Khorana HG. Studies on polynucleotides. XXIV. The stepwise synthesis of specific deoxyribopolynucleotides (4). Protected derivatives of deoxyribonucleosides and new syntheses of deoxyribonucleoside-3′-phosphates J Am Chem Soc. 1963; 85: 3821-7.
- Yantsevich A, Shchur VV, Usanov SA. Oligonucleotide Preparation Approach for Assembly of DNA Synthons. SLAS technology. 2019; 24: 556-68.
- Shchur VV, Burankova YP, Shapira MA, Klevzhits DV, Usanov SA. et.al. Programmed assembly of long DNA synthons: design, mechanism, and online monitoring. Applied microbiology and biotechnology. 2019; 103: 9103-17.
- Dellinger DJ, Wyrzkewicz TK, Betley JR and Caruthers MH. Solid-phase synthesis of oligodeoxyribonucleotides using phosphoramidite synthons in a two step synthesis cycle, Poster 209, XIIIth International Round Table: Nucleosides, Nucleotides and their Biological Applications, Montpellier. 1998.
- Stemmer W P, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides Gene. 1995; 164: 49-53.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. et.al. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004; 25: 1605-12.
- Zhao S, Li J, Wang L, WangX. Degradation of Rhodamine B and Safranin-T by MO3CeO2 Nanofibers and Air Using a Continuous Mode. Clean-Soil Air Water 2010; 38: 268-274.