
Review Article
Austin Chem Eng. 2022; 9(1): 1089.
Design and Synthesys of Highly Stretchable and Tough Adhesive Hydrogels for Dry and Wet Biomedical Applications
Neves D, Oechsler BF and Machado RAF*
Chemical Engineering Program, Federal University of Santa Catarina, Florianopolis, Santa Catarina State, Brazil
*Corresponding author: Ricardo AF Machado, Chemical Engineering Program, Federal University of Santa Catarina, PO Box 476, Zip Code 88040900, Florianopolis, Santa Catarina State, Brazil
Received: March 22, 2022; Accepted: April 26, 2022; Published: May 03, 2022
Abstract
The development of less invasive procedures and devices in the biomedical applications of wound closure and healing is an important and essential task. The healing process is facilitated by proper closure of the surgical wound; traditionally a particularly invasive practice. Tissue adhesives design should incorporate simplicity, safety, and painless removal from the skin. Moreover, these materials should have adequate adhesive strength, be tough enough to incorporate the mechanical skin behaviors, and must also show compatibility with body fluids as well as cells and tissues. The design of tough adhesives (TAs) can potentially enable many applications, including the gluing of tissues and attaching devices in vivo, tissue repair, and can be used as a hemostatic dressing because of their compatibility with blood exposure. This work reviews many strategies to design tough hydrogels with the introduction of non-covalent bonds and the construction of stretchable polymer networks and interpenetrated networks, such as a double-network hydrogel. To overcome such difficulties, some synthetic wet adhesives emulating natural adhesive materials of marine organisms have been investigated.
Keywords: Tough Adhesive Hydrogels; Highly Stretchable Adhesives; Dry and Wet Biomedical Applications
Introduction
Tissue adhesives are used as an alternative to stitches or staples and can be less damaging to the healthy tissues, for example in sutureless in wound closure and wound dressings [1,2]. However, these materials can suffer from low biocompatibility and poor matching of the mechanical properties with the tissues. Therefore, compatibility with body fluids, cells, and tissues is a desirable property [3]. This family of adhesives may be useful in several biomedical applications, including tissue adhesives, wound dressings, and tissue repair.
Adhesion to wet surfaces, including biological tissues, is important in many fields but it has proven to be extremely challenging. Existing adhesives are cytotoxic, adhere weakly to tissues, or cannot be used in wet environments. Firstly the adhesive should form a strong bond with the substrate and, second, the material inside either the adhesive or the substrate should dissipate energy by hysteresis [4].
Commercial strong cyanoacrylate adhesives are incompatible with wet surfaces as they solidify immediately in contact with water, forming a rigid structure that cannot be flexible to the tissue movements, and they exhibit cellular toxicity [5,6]. Biohybrid polymers can be an advanced way that combines properties of adhesiveness, biodegradability, absorbency, and stretching force [7]. Hydrogels have immense potential as reversible wet adhesives when they are integrated with biomimetic microstructures, in which some factors can be considered. Firstly, the hydrophilic nature of the hydrogels is beneficial to induce strong capillary adhesion under wet conditions. Second, they can absorb a large quantity of water at the contact interface, which would allow closer contact with the target substrate [8,9].
While several promising properties and multiple functionalities, such as biocompatibility and environmental friendliness, responsiveness to external stimuli, adhesion, anti-biofouling behavior, and biodegradability have been integrated into hydrogels, the relatively poor mechanical behavior of hydrogels remains a challenge, impeding their use in real-world applications that require mechanical integrity. Particularly, they can be designed to achieve better physical, chemical, electrical and biological properties [10,11]. Furthermore, they are biocompatible and therefore can be used for diverse biomedical applications such as tissue adhesives, tissue repair, scaffolds for cell growth, and wound dressings. Indeed, in recent years, hydrogels have been actively explored as materials for efficient wet adhesives [3,12].
Hydrogels with a single component have a poor mechanical property [13], however, several recent reports related below have shown an improvement in the toughness from the selection of hybrid or composite hydrogels with non-covalent bonds (e.g., hydrogen bonds and ionic interactions), highly stretchable networks and double-network.
In 2003, Gong et al. [11] discovered a double-network highly stretchable hydrogel, and San et al. [14] synthesized a highly stretchable and tough hydrogel, prepared from a network of polyacrylamide and alginate. Ballance et al. [15] demonstrated that linking cyclodextrin chemically in a stretchable polyacrylamide hydrogel increases mechanical strength. Stretching these hydrogels loaded with quinine significantly enhanced the antibacterial activity of the gels due to the increased amount and release rate of quinine, demonstrating antibacterial behavior.
Tough Hydrogels - Dissipative Matrices
Hydrogels are materials that consist of crosslinked polymer networks dispersed in water [16]. Most common hydrogels used for wound dressings or drug delivery systems are brittle, not exhibiting high stretching resistance due to their mechanical behavior. In recent years, intense efforts have been directed toward designing synthetic hydrogels possessing high strength and toughness by engineering energy- dissipative pathways into the gel structure [17,18].
In order to expand the scope of functional hydrogel materials, there is a need to develop new mechanically strong and tough supramolecular hydrogels that could not only be easily accessed from simple and commercially available starting materials, but that also exhibit a combination of desired mechanical properties such as high strength, stretchability, self-recovery, self-healing, fatigue-resistance, and thermoplasticity [19].
Hydrogels with a double or more network of two or more polymer chains, crosslinked separately, in which one of them having short chains and the other having long chains can enable mechanisms to dissipate energy and became tougher [14,20-22].
These double network hydrogels have been demonstrated synergistically combine the stiffness of a tightly cross-linked network with the dissipative ability of an independently formed, loosely crosslinked network. However, the exceptional toughness of a double network gel compromises the material’s fatigue resistance when stretched, as it depends entirely upon the permanent rupture of the short-chain as an energy dissipation mechanism, while the long chains assist in broadening the fracture zone to maximize dissipation. Once the first network has been damaged, the mechanical response is dominated by the much softer second network, and the high stiffness and toughness are lost [14,20]. One way to replace this situation for recoverable energy dissipation is to change the covalent bonds to noncovalent bonds [20].
Li et al. [4] created a bioinspired design for an adhesive consisting of two layers: an adhesive surface with a flexible dissipative matrix, inspired by defensive mucus secreted by slugs that strongly adheres to wet surfaces. The adhesive layer adheres to the substrate by electrostatic interactions, covalent bonds, and physical interpenetration. The second layer is composed of a matrix of hydrogel that can amplify energy dissipation through hysteresis. The two layers lead to higher adhesion energies on wet surfaces when compared with existing adhesives. This bioinspired adhesive has an ideal level of the stick and moves along with the living tissue.
Sun et al. [14] developed the first highly stretchable and tough hydrogels with better deformation and energy dissipation with enhanced mechanical properties, mixing two types of crosslinked polymers: ionic crosslink alginate and a covalently cross-linked polyacrylamide. Alginates chains consist of mannuronic acids (M block) and guluronic acids (G blocks), isolated in their chains or alternated blocks. In water solution, the G blocks on different chains form ionic crosslinks through divalent cations (Ca2+), resulting in a network in water, an alginate hydrogel. The polyacrylamide chains in hydrogel form a network by covalent crosslinks (Figure 1).

Figure 1: a) Alginate Hydrogel. M blocks and G blocks forming ionic crosslinks through Ca2+. b) Polyacrylamide hydrogel chains formed by covalent crosslinks
through N,N-methylenebisacrylamide (MBAA). c) A hybrid alginate- polyacrylamide hydrogel with two types of networks. Adapted from Sun et al. [14].
Sun et al. [14] stretched the alginate-polyacrylamide hybrid hydrogel over 20 times its original length without rupture. The stress and the elongation at rupture were 156kPa and 23cm respectively, against 3.7kPa and 1.2cm for the alginate gel and 11kPa and 6.6cm for the polyacrylamide gel. When the proportion of acrylamide was increased, the elastic modulus of the hybrid hydrogel was reduced and the critical elongation at rupture reached the maximum when acrylamide was 89 wt.%. When the hybrid gel is stretched, the polyacrylamide network remains intact and stabilizes the deformation, while the alginate network unzips progressively, with closely spaced ionic crosslinks unzipping at a small stretch, followed by more and more widely spaced ionic crosslinks unzipping as the stretch increases.
Yuk et al. [23] reported a design strategy and a set of simple fabrication methods to give extremely tough and functional bonding between hydrogels content over 90 wt.% of water and diverse solids like glass, silicon, ceramics, aluminum, and others. These materials do not require porous or topographic surfaces, achieving interfacial toughness values over 1,000J/m².
The proposed strategy designed for the tough hybrid hydrogelsolid bonding is illustrated in Figure 2, as interfacial cracks can kink and propagate in relatively brittle hydrogel matrices; the design of tough hydrogel-solid bonding first requires high fracture toughness of the constituent hydrogels. Part “a” shows that the tough bonding first requires high fracture toughness of the constituent hydrogels, whereas tough hydrogels generally consist of long- chain polymer networks and mechanically dissipative components. This is sufficient to achieve tough bonding by chemically anchoring the long-chain networks on solid surfaces. In part “b”, the chemical anchoring gives a relatively high intrinsic work of adhesion G0, which maintains the cohesion of the hydrogel-solid interface and allows large deformation and mechanical dissipation to be developed in the hydrogel during detachment. The dissipation further contributes to the total interfacial toughness by GD. Part “c” shows a schematic of hydrogel–solid interface [23].
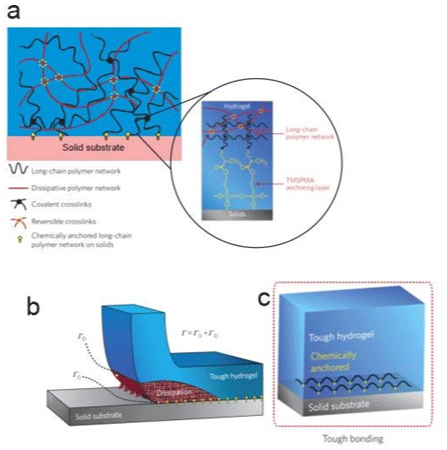
Figure 2: A design strategy for tough bonding of hydrogels to diverse solids. a) Long-chain polymer network anchored by a crosslinked network between the
functional silane onto solids surface. b) Chemical anchoring with a high intrinsic work of adhesion G0; c) A schematic of hydrogel-solid interface. Adapted from Yuk
et al. [23].
Whereas tough hydrogels generally consist of covalently crosslinked long-chain polymer networks that are highly stretchable and other components that dissipate mechanical energy under deformation, it is impractical to chemically bond all components of the hydrogels on solid surfaces [17,24].
Yuk et al. [23] propose that it is sufficient to achieve relatively tough hydrogel-solid bonding by chemically anchoring the longchain polymer network of a tough hydrogel on solid surfaces (Figure 2a.). Then, when the hydrogel chemically anchored is peeled off, the separation of the anchored layer of polymer chains gives an intrinsic force of adhesion, G0, while the hydrogel around this scission region will be deformed and will dissipate a lot of mechanical energy [25] that can contribute for the interfacial toughness by GD. The design strategy of Yuk et al. [23] is to use a functional silane, 3-(trimethoxysilyl) propyl methacrylate (TPMSPMA) to modify the solid surface like in Figure 2d.
Yuk et al. [23] covalently crosslinked the long-chain polymers network of polyacrylamide (PAAm) or polyethylene glycol diacrylate (PEGDA) to the silanes on the modified surfaces of various solids. To form tough hydrogels, the long-chain polymer network is interpenetrated with an irreversibly crosslinked network of alginate, chitosan, or hyaluronan, in which the reversible crosslinking and chain scission dissipate mechanical energy (Figure 2a and 2b). The peel adhesion force (90° degrees peeling test) was carried out with a peeling rate of 50mm/min to measure the interfacial toughness between the hydrogel (3mm thicker) and a glass substrate. The initial measure of interfacial toughness was 24J/m2 (Figure 3b) than a crack initiates at the interface. As the peeling force increases, the hydrogel around the interfacial crack becomes highly deformed and unstable. During the crack propagation, the bonds increasing in amplitude and form wavelengths until to detach from the substrate (Figure 3c), and in this stage, the measured interfacial toughness was over 1,500J/ m². Different results were fitted to different peeling rates, decreasing with lower rates, so it is evident that the measure of toughness chemically anchored hydrogel is rate dependent, possibly owing to the viscoelasticity of the hydrogel.
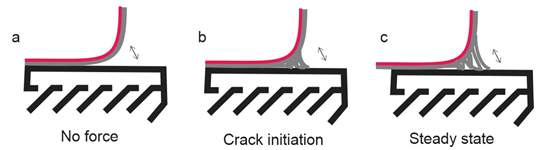
Figure 3: The peeling process of the tough hydrogel chemically anchored on the glass substrate.
Yuk et al. [23] also test the PAAm-alginate hydrogel on substrates in water for 24h, and the measures of interfacial toughness values for swollen hydrogels bonded on glass were 1,123J/m², that are consistently high, indicating that this strategy can produce tough hydrogel to diverse solids on the wet environment.
Jeon et al. [26] synthesized an extremely stretchable and fast selfhealing hydrogel, which can stretch up to 100 times their initial length and self-heal within the 30s, using a novel crosslinker consisting of an acrylic head, a hydrophobic alkyl spacer connected by carbamate, and 2-ureido-4-pyrimidone tail (UPyHCBA) (Figure 4a). The 2-ureido-4-pyrimidone (UPy) tail forms self-complementary dimers with quadruple H-bonds. The micellar polymerization was carried out with sodium dodecyl sulfate (SDS) micelles as a hydrophobic environment for the UPyHCBA and acrylamide (Figure 4b-4d). The results are hydrogels with approximately 81 wt% of water, stretching more than 100 times, quite soft, and not so easy to fracture. Both the elastic strength and maximum stress increase with higher UPyHCBA concentration, while the strain to fracture decreases.
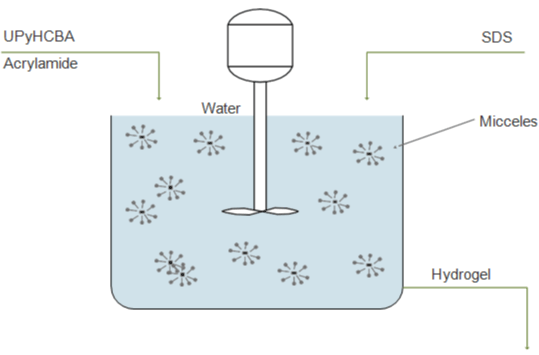
Figure 4: Copolymerization of UPyHCBA and acrylamide schema.
To characterize the self-healing behavior of the gels, Jeon et al. [26] cut samples into two different pieces, after this, they were held together for 3min, positioned in the tensile tester. The healing sample showed an elevated stress level at small stretches and demonstrated that can heal quickly and form bonds that are at least as strong as the initial gel. It was observed that decreasing the SDS concentration, the tackiness increases by releasing the UPy units outside of the micelles, and the ability to self-heal decreases significantly (Figure 5). This ability of this gel stems from the formation of intermolecular H-bonds across the damaged interface.
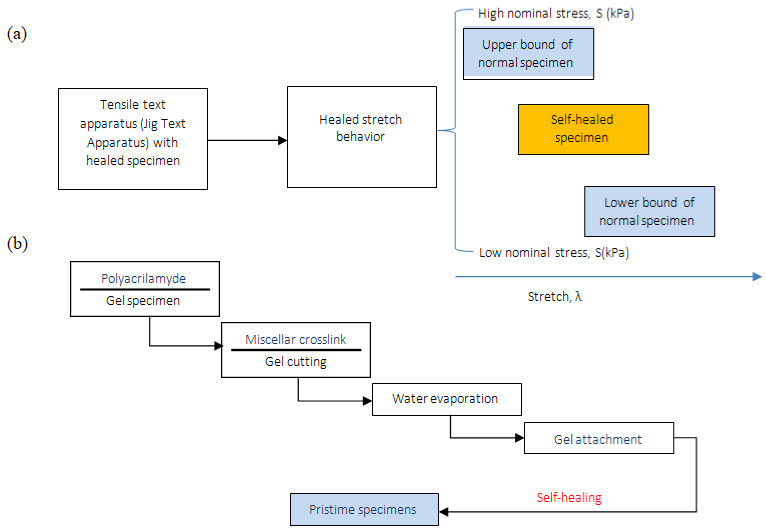
Figure 5: (a) Tensile test schema. (b) Self-healing schematic representation.
A tough hydrogel that can be stretched up to ≈30 times its original length, self-healing due to the interchain hydrogen bonds between polyacrylamide and chitosan was synthesized by Dutta et al. [19]. They incorporated short chain chitosan (CS) molecules (4.7 wt %) into a physical (hydrogen-bonded) gel network of polyacrylamide (PAM) and in the absence of a covalent cross-linker. These gels show thermoplastic behavior and can be molded into different shapes after a heating-cooling cycle, maintaining reasonable mechanical strength. The gels can withstand repeated compressive loading-unloading cycles, exhibiting reliable load-bearing capacity. Beyond the hydrogen bonds of PAM-CS cross-links, believes that a synergistic with physical entanglement of PAM and CS chains can be responsible for enhanced mechanical properties too. Besides, an elevated concentration of CS, the CS-CS hydrogen- bonding interactions may compromise the PAM-CS interchain hydrogen bond formation, leading to loss of mechanical properties.
Dutta et al. [19] investigated the behavior of the hybrid hydrogel in body fluids environment to apply in tissue engineering. After 1h immersed in water, the hybrid hydrogel PAM-CS sustained ~130kPa of stress after ten consecutive cycles of the compression-relaxation test at 90% compressive strain, when compared to ~300kPa with the same hydrogel not immersed. The hybrid hydrogel swelled when immersed in water, keeping its shape until ~6h, higher than the control PAM hydrogel which loses its shape within ~15min when immersed. Even with the weakening of gel strength after swell, and when submitted for consecutive cycles of compression-relaxation, this gel was still not broken and was able to return to the original shape.
A series of hyperbranched polysiloxane (HSi) with tunable molecular structure, highly cross-linked polymer network, was designed by Li et al. [27], synthesized, and used as an efficient crosslinker to get a polyacrylamide/chitosan (PCH) hydrogel with high strength and toughness. The obtained HSi with bi-functional groups endowed PCH hydrogels presented mechanical properties better than HSi containing single-functional group. Furthermore, the resulting hydrogels also showed an outstanding anti-fatigue ability to withstand cyclic compressive tests. Tensile strength of 302kPa, elongation at break of 2263%, and toughness of 3,850J/m² can be achieved by optimizing the molecular structure and content of HSi with a bi-functional group. The improved mechanical properties are likely derived from the combination of homogeneous and strong polymer networks and improved interactions of polymer chains (e.g. covalent and hydrogen bonds) [27].
In summary, hybrid hydrogels dissipate energy effectively, showing pronounced hysteresis, which can decreases if the load and unload are applied immediately [14]. The rupture energy of the hybrid gel is much larger than other tough synthetic gels reported.
Tough Hydrogels - Adhesives Matrices
Commercial biocompatible adhesives can be vulnerable to debonding depending on the type and force of the bond formed with the substrate, and the energy dissipated by hysteresis. Cyanoacrylates (CAs) are a strong tissue adhesive but it is cytotoxic and deteriorates and solidifies in contact with wet surfaces and tissues. To overcome such difficulties, there are many studies in synthetic wet adhesives emulating natural adhesive materials of marine organisms as described below.
Marine mussels can adhere to diverse wet and dry surfaces because of a series of adhesive proteins [28]. Li et al. [4] reported a tough adhesive made of an adhesive layer containing an interpenetrating positively charged polymer, that can bond onto the substrate through electrostatic interactions, covalent bonds, and physical interpenetration, and a second matrix layer that can dissipate energy through hysteresis under deformation. Here there is a 2-component junction, a tough hydrogel with possibilities already commented in the previous topic, with an adhesive. The criteria were: 1) The adhesive must form covalent bonds across the interface on wet negatively charged surfaces of tissues and cells, inspired in slug adhesive, such a polymer can be absorbed to the surface via electrostatic attractions, enabling primary amine groups to bind covalently with carboxylic acid groups from the hydrogel matrix and the tissue surface, and, 2) The matrix hydrogel layer must be tough and dissipate energy under stress forces, like the alginate-polyacrylamide (Alg-PAAm).
A family of tough adhesives that can be adhered to wet surfaces with many compositions was developed by Li et al. [4]. The best performance was obtained with chitosan as a bridging adhesive polymer with two reagents, 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide and N-hydroxysulfosuccinimide, to facilitate the covalent bond formation, and Alg-PAAm hydrogel inspired in Sun et al. [14] (Figure 6a-6d), that effectively dissipates energy under deformation. Analyzing the peel adhesion on porcine skin, the toughadhesive (TA) led >1000J.m², according to Figure 6e, and exhibited a rapid increase in adhesion energy over time. When the porcine skin was covered by whole blood, the energy of TA was found to be 1116 J.m2, strong and very similar with the dry skin, and CA demonstrates deterioration on the wet surface, around 100 J.m² of adhesion force [4].
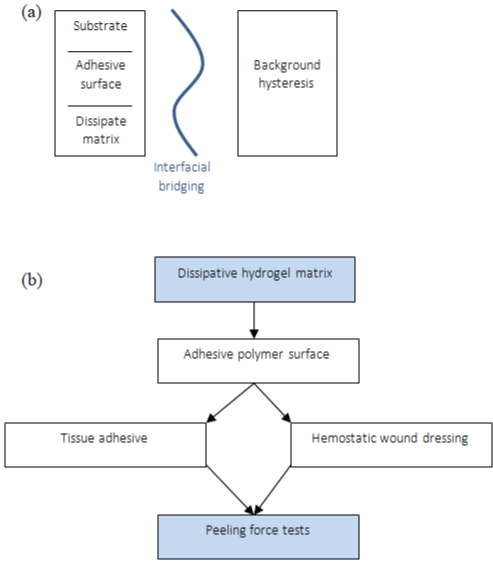
Figure 6: (a) Schematic of dissipative hydrogel matrix with an adhesive polymer surface, (b) Peeling force tests: Application of tough adhesive (TA) and
cyanoacrylate (CA).
Zhang et al. [29] developed a hyperbranched adhesive that, although not moisture sensitive, would function in wet conditions and overcome the current drawbacks of cyanoacrylates (toxicity) and fibrin adhesives (poor strength). In their study, they compare a marine mussel adhesive that functioned with strong adhesiveness in wet conditions and low cytotoxicity, polymerizing trimethylolpropane triacrylate with dopamine hydrochloride and dimethyl sulfoxide by polycondensation, through Michael addition reaction (Figure 7), obtaining a 3D hyperbranched polymer dopamine co-acrylate (PDA) with many vinyl groups, reaching 37kPa in just 15 minutes when fibrinogen was used as curing agent.
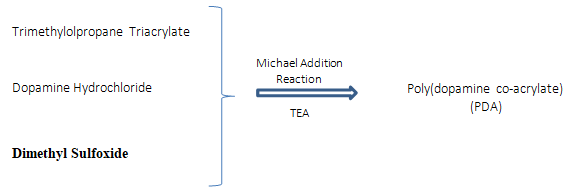
Figure 7: Reaction mechanism of self-condensation between dopamine and triacrylate monomers through Michael Addiction reaction.
Dopamine has been used to improve the adhesive property of hydrogel, especially in wet conditions. This adhesive force in few minutes is very substantial for incisional skin wound closure surgery applications, while a fibrin adhesive yielded mean lap shear strength of 9.45kPa and 15.34kPa after 1h and 1-day curing time respectively. Fibrinogen proved to be a good curing agent achieving high adhesion in a short time, but for a long period of curing. Horseradish Peroxidase (HRP) achieves the highest adhesion force (76 ±13.4 kPa) after one day curing period, more than the fibrinogen, which achieved around 60kPa after one day of curing time. Otherwise, HRP achieves around 20kPa in 15min of curing time [29].
According to Ishida et al. [30], hyperbranched polymers are not difficult to carry out in one-pot polymerization by one-step polycondensation, with two different functional groups ABn (n≥2), reacting with each other and not undergoing a self reaction. Usually, direct polycondensation of A2 (Bifunctional monomer) and B3 (tri-functional monomer) monomers generally results in gelation. However, soluble three-dimensional hyperbranched polymers as the PDA were obtained by tightly controlling the concentration, the ratio of A2 and B3 monomers, the reaction time, and stopping the polymerization via end-capping before the critical point of gelation [29].
Based on mussel’s adhesion mechanisms (Figure 8), a two-step process aiming to develop an adhesive and tough polydopamine-claypolyacrylamide (PDA-clay-PAM) adhesive hydrogel was proposed by Han et al. [1].

Figure 8: Polymerization design for the preparation of PDA-clay-PAM hydrogel.
The synthesized hydrogel presented repeatable and durable adhesiveness on the skin without damage and easy removability. The secret is to maintain enough catechol groups in the hydrogel. Firstly, dopamine (DA) was intercalated into layers of clay nanosheets where its oxidation was limited in the confined nanospace, which is a structure with free catechol groups. Next, acrylamide monomers (AM) were added and polymerized in situ by free radical polymerization with initiator and cross-linker (Figure 8), forming a free-standing and adhesive PDA-clay- PAM hydrogel. Incorporating polydopamine into polyacrylamide (PAAm) covalently crosslinked network gives the hydrogel with adhesive property to human skin [1].
The superior mechanical properties of the PDA-clay-PAM hydrogel were attributed to the PDA-intercalated clay nanosheets serving as functional nanofillers to interact with the polymer network to reinforce the hydrogel. Furthermore, the catechol groups have been incorporated to toughen hydrogels and help PDA chains to interact with the polymer networks through bonding noncovalent to consume energy effectively during the deformation process [1,31]. Incorporating catechol into non-adhesive synthetic (e.g. poly(ethylene glycol) (PEG), acrylate, and acrylamide polymer) and biological polymers (e.g. gelatin, chitosan, and alginate) has enabled these materials to bond to tissue covalently and non-covalently [31].
The hydrogel exhibited adhesion to the merged glass slides, natural surfaces such as hydrophilic rock, hydrophobic leaves, and fresh organ tissues with tissue fluid. Moreover, high adhesion strength to glass, titanium (Ti), polyethylene (PE), and porcine skin was seen, and the results were 120KPa, 80.8KPa, 80.7KPa, and 28.5KP, respectively (Figure 9a). The hydrogel did not adhere to surfaces under aqueous conditions. Qu et al. [32] reached adhesion strength on porcine skin about 4.4kPa to 6.1kPa by addition of Pluronic®F127 in a series of a hydrogel, and one can see much lower adhesion strength than ones presented by Han et al. [1]. However, the strong adhesion reported by Han et al. [1] was immediately recovered when water was removed from the hydrogel surfaces. Amazingly, the PDA-clay-PAM hydrogel was able to adhere to human skin with no irritation and could be removed easily without causing any damage or pain (Figure 9). The PDA-clay-PAM hydrogel exhibited repeatable and durable adhesion demonstrated by performing an adhesion peeloff test on porcine skin over 20 cycles and could be stretched to 20 times its initial length and recovered to its initial length after 10 min of storage, recovering to its original shape after it was compressed up to the strain of 80% [1].
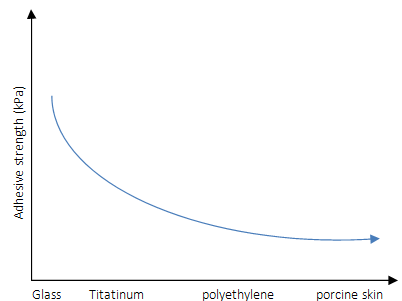
Figure 9: Adhesive strength schema of the hydrogels to porcine skin tested by tensile adhesion tests.
The better mechanical properties are ascribed to the presence of covalent and noncovalent cross-linked polymer networks and clay nano reinforcements. Data of vitro cell culture indicated that the adhesive and tough hydrogel favors cell adhesion and proliferation, owing to the PDA component full-thickness skin defect repair experiments. These results showed that the hydrogel could accelerate wound healing, demonstrating that the inclusion of PDA is a feasible method for developing a tough cell and tissue-adhesive hydrogel, including skin regeneration and biocompatible product [1]. Another synthesis of PDA self-healing and stretchable hydrogel with enhancing adhesiveness was reported by He et al. [33], from the introduction of a microgel into the hydrogel matrix of polydopamine (poly (N-isopropyl acrylamide) microgel/polyacrylic acid-polyacrylamidepolydopamine (MR/PAAc-PAM-PDA) hydrogel).
Inspired by the contraction ability of embryonic wounds, Blacklow et al. [34] presented a new biomechanical adhesive wound dressing, called active adhesive dressings (AADs), consisting of a thermoresponsive tough adhesive hydrogel, with high stretchable, tough, and antimicrobial properties. In contact with the wounds, it responses to the skin temperature and contract the wound edges together (Figure 10).

Figure 10: Bioinspired design of AAD for promoting wound contraction, enabled by AAD that adheres to the wound and contracts its edges at the skin temperature.
Red dashed arrows indicate the contraction.
Blacklow et al. [34] used the same method as Li et al. [4] to obtain a tough adhesive hydrogel, combining elements like a thermoresponsive polymer, as poly(N-isopropyl acrylamide) (PNIPAm), and silver nanoparticles (AgNPs) to obtain a thermoresponsive behavior at around 32°C and antimicrobial function respectively (Figure 11). The in vitro and in vivo studies demonstrated that the AAD actively contracted wounds and accelerated wound healing.
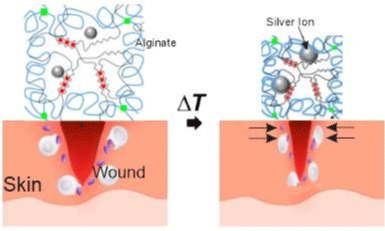
Figure 11: Schematics of the temperature-triggered transition of AAD, which consist of PNIPAm (blue lines), alginate (black lines), and AgNPs (Ag; grey spheres).
AAD forms strong adhesion (green lines) with the wounded skin. Adapted from Blacklow et al. [34].
A hybrid network was formed at 4°C (HN) and exhibited high toughness around 500J.m² (shear test to quantify the toughness by fracture energy), whereas the single-network alginate (Alg) or PNIPAm hydrogels (SN) exhibited fracture energy lower than 100J. m2 (Figure 12). The inclusion of AgNPs had no significant impact on the matrix toughness. With increasing concentration of NIPAm monomers, the fracture energy was further increased beyond 2000J.m-2. The results confirmed the synergistic effect of the PNIPAm and alginate networks for high mechanical performance. Adhesion peel strength demonstrates that hydrogels embedded AgNPs HNAg achieved high adhesion on porcine skin (175J.m²), slightly higher than HN (125J.m²), and substantially higher than dressings used clinically and Band-Aid® (Figure 12) [34].
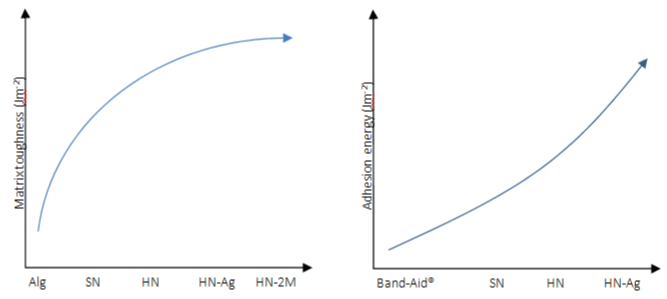
Figure 12: Matrix toughness and adhesion energy on porcine skin of various hydrogel matrices. Alg refers to alginate hydrogels and 2M refers to 2M NIPAm
monomers used to form HN.
In vitro experiments demonstrated that AAD can apply contractile strains to wound edges on the skin following temperature-triggered contraction. The results showed that AAD reduced the wound area to around 45%, compared to minimal changes in the non-treatment control and the TA condition. When applied AAD in vivo rodent skin wounds, a phase transition of the AAD occurred at 40min and significantly accelerated the wound closure as early as day 3, with a significant improvement on day 7. In vitro tests of AAD-enabled wound contraction on fresh rodent skin. Figure 13b shows digital images of the initial wounds and those after 7 days with no hydrogel treatment (control), with treatment with nonthermoresponsive TA, and with treatment with AAD, with scale bars of 2mm.
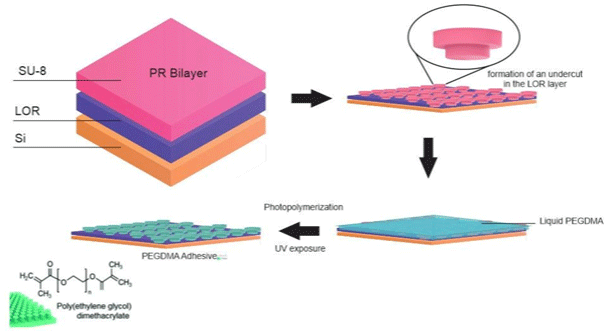
Figure 13: Schematic of the fabrication procedure of the PEGDMA adhesive.
A bioinspired wet adhesive hydrogel-based, which can exhibit immediate, strong, and reversible adhesion under wet and underwater conditions, was developing by Yi et al. [35]. Under dry conditions, the microstructures of this hydrogel exhibit strong Van der Waals interaction-based adhesion, while under an underwater environment; it can maximize capillary adhesion, based on the water-absorbing properties of the microstructured hydrogel. A bioinspired micropillar array based on polyethylene glycol (PEG) hydrogel, which can absorb a large quantity of water and is hydrophilic, allowing the contact interface by capillary adhesion and conformal physical contact to target surfaces (Figure 13), was synthesized by Yi et al. [35].
An ultratough and self-healing hydrogel with high cell affinity and tissue adhesiveness is prepared by Chen et al. [36], based in a double-network of dopamine grafted oxidized sodium alginate (OSA-DA) and polyacrylamide (PAM), reaching highly self-healing behavior (80% of the mechanical recovery in 6h), high tensile strength, ultra-stretchability (2550%) and highly adhesive strength on diverse surfaces. Firstly dopamine was grafted to the oxidized sodium alginate, forming dopamine-grafted oxidized sodium alginate (OSA-DA), then, acrylamide monomers were joined chemically polymerized, forming OSA-DA-PAM hydrogel. Dynamic covalent cross-linking was formed through Schiff base reaction between the aldehyde groups of the OSA-DA chains and the amino groups of the PAM chains. Reversible hydrogen bonds were formed between catechol groups in the OSA-DA chains. Hence, the double network hydrogel was physically and chemically cross-linked (Figure 14) [36].

Figure 14: Synthetic process and schematic structure of OSA-DA-PAM hydrogel chemically polymerized in presence of ammonium persulfate (APS) and N,
N-methylenebis-acrylamide (BIS).
The hydrogel was cut into two halves and then placed into physical contact again for 1h. When two halves were placed into contact, dynamic Schiff bonds between OSA-DA and PAM functional groups re-formed, leading to the recovery of the hydrogel. After 6h, the tensile strength and fracture strain of the self-healed OSA-DA-PAM-3 hydrogel can be recovered significantly. The self-healing efficiency, defined as the tensile strength ratio of the self- healed sample to the original sample, was 80% for the OSA-DA-PAM-3 hydrogel [36].
It is clear that there is a transformative advance in achieving strong hydrogel adhesion, which is a supramolecular phenomenon. The design of smart surfaces with switchable adhesive properties in a wet environment has remained a challenge in adhesion science and materials engineering, in synthesize bioinspired adhesive, which entails the reversible, tunable, and fast regulation of the wet adhesion on diverse surfaces. It is necessary to be taken into consideration in designing tissue engineering matrices by polymerization, as is the risk of cytotoxicity linked to unreacted monomers. For example, PAAm is popular for tissue engineering, however, the AAM monomer is a known neurotoxin and a risk factor for several cancers [37,38].
We know that strong hydrogel adhesion requires the synergy of chemistry of bonds, topology of connection, dissipation and mechanics. This synergy characterizes a fundamental hydrogel adhesion to various materials and applications especially in wet environments, including wound closure, tissue repair, drug delivery systems, medical instruments, biomedical devices, etc.
Concluding Remarks
A tough hydrogel adhesive (bioinspired or not) with smart adhesion performance can be used as a versatile and advanced adhesive for various biomedical applications that stable and repeatable adhesion under diverse dry and wet environments is necessary. Approaches to the fabrication of stretchable and tough hydrogels include crosslinking two polymers or mixing one polymer with micelles or nanomaterials. Highly stretchable and tough hydrogels, with great adhesion strength and healing times, can be utilized for wound healing.
Many tough hydrogels bring new functions, but mass production is hampered by the lack of established methods for fabrication.
References
- Han Lu, Lu Xiong, Liu Kezhi, Wang Kefeng, Fang Liming, Weng Lu-Tao, et al. Mussel-Inspired Adhesive and Tough Hydrogel Based on Nanoclay Confined Dopamine Polymerization. ACS Nano. 2017; 11: 2561-2574.
- Han L, Lu X, Wang M, Gan D, Deng W, Wang K, et al. A Mussel- Inspired Conductive, Self-Adhesive, and Self-Healable Tough Hydrogel as Cell Stimulators and Implantable Bioelectronics. Small. 2017; 13.
- Jing X, Mi H-Y, Lin Y-J, Enriquez E, Peng X-F, Turng L-S. Highly Stretchable and Biocompatible Strain Sensors Based on Mussel-Inspired Super- Adhesive Self-Healing Hydrogels for Human Motion Monitoring. ACS Appl. Mater. Interfaces. 2018; 10: 20897-20909.
- Li J, Celiz AD, Yang J, Yang Q, Wamala I, Whyte W, et al. Tough adhesives for diverse wet surfaces. Science. 2017; 357: 378-381.
- Fortelny RH, Petter-Puchner AH, Walder N, Mittermayr R, Öhlinger W, Heinze A, et al. Cyanoacrylate tissue sealant impairs tissue integration of macroporous mesh in experimental hernia repair. Surg. Endosc. 2007; 21: 1781-1785.
- Vakalopoulos KA, Wu Z, Kroese L, Kleinrensink GJ, Jeekel J, Vendamme R, et al. Mechanical strength and rheological properties of tissue adhesives with regard to colorectal anastomosis: An ex vivo study. Ann. Surg. 2015; 261: 323-331.
- Liu Y, Meng H, Konst S, Sarmiento R, Rajachar R, Lee BP. Injectable Dopamine-Modified Poly(ethylene glycol) Nanocomposite Hydrogel with Enhanced Adhesive Property and Bioactivity. ACS Appl. Mater. Interfaces. 2014; 6: 16982-16992.
- Hoffman AS. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012; 64: 18-23.
- Ghobril C, Grinstaff M. The chemistry and engineering of polymeric hydrogel adhesives for wound closure: A tutorial. Chem. Soc. Rev. 2015; 44: 1820- 1835.
- Gaharwar AK, Peppas NA, Khademhosseini A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 2014; 111: 441-453.
- Gong JP, Katsuyama Y, Kurokawa T, Osada Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003; 15: 1155-1158.
- Darnell MC, Sun J-Y, Mehta M, Johnson C, Arany PR, Suo Z, et al. Performance and biocompatibility of extremely tough alginate/polyacrylamide hydrogels. Biomaterials. 2013; 34: 8042-8048.
- Kamoun EA, Kenawy E-R. A Review on Polymeric Hydrogel Membranes for Wound Dressing Applications: PVA-based Hydrogel Dressings. Journal of advanced research. 2017; 8: 217-233.
- Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, et al. Highly Stretchable and Tough Hydrogels. Nature. 2012; 489: 133-136.
- Ballance WC, Seo Y, Baek K, Chalifoux M, Kim D, Kong H. Stretchable, antibacterial hydrogel activated by large mechanical deformation. J. Control. Release. 2018; 275: 1-11.
- Lee KY and Mooney DJ. Hydrogels for Tissue Engineering. Chemical Reviews. 2001; 101: 1869-1879.
- Zhao X. Multi-scale multi-mechanism design of tough hydrogels: Building dissipation into stretchy networks. Soft Matter. 2014; 10: 672-687.
- Zhen Qiao, Meijuan Cao, Kathryn Michels, Lee Hoffman, Hai-Feng Ji. Design and Fabrication of Highly Stretchable and Tough Hydrogels, Polymer Reviews. 2019.
- Dutta A, Maity S, Das RK. A Highly Stretchable, Tough, Self-Healing, and Thermoprocessable Polyacrylamide-Chitosan Supramolecular Hydrogel. Macromol. Mater. Eng. 2018: 1800322.
- Henderson KJ, Zhou TC, Otim KJ, Shull KR. Ionically cross-linked triblock copolymer hydrogels with high strength. Micromolecules. 2010; 43: 6193- 6201.
- Sanabria-DeLong N, Crosby AJ, Tew GN. Photo-Cross-Linked PLA-PEO-PLA Hydrogels from Self- Assembled Physical Networks: Mechanical Properties and Influence of Assumed Constitutive Relationships. Biomacromolecules. 2008; 9: 2784-2791.
- Branco MC, Nettesheim F, Pochan DJ, Schneider JP, Wagner NJ. Fast Dynamics of Semiflexible Chain Networks of Self-Assembled Peptides. Biomacromolecules. 2009; 10: 1374-1380.
- Yuk H, Zhang T, Lin S, Parada GA, Zhao X. Tough bonding of hydrogels to diverse non-porous surfaces. Nat. Mater. 2016; 15: 190-196.
- Gong JP. Why are double network hydrogels so tough? Soft Matter. 2010; 6: 2583-2590.
- Derail C, Allal A, Marin G & Tordjeman P. Relationship between viscoelastic and peeling properties of model adhesives. Part 1. Cohesive fracture. J. Adhes. 1997; 61: 123-157.
- Jeon I, Cui J, Illeperuma WR, Aizenberg J and Vlassak JJ. Extremely Stretchable and Fast Self-Healing Hydrogels. Adv. Mater. 2016; 28: 4678- 4683.
- Li SN, Li B, Gong LX, Yu ZR, Feng Y, Jia D, et al. Enhanced mechanical properties of polyacrylamide/chitosan hydrogels by tuning the molecular structure of hyperbranched polysiloxane, Materials & Design. 2019; 162: 162-170.
- Ahn BK. Perspectives on Mussel-Inspired Wet Adhesion. J. Am. Chem. Soc. 2017; 139: 10166-10171.
- Zhang Hong, Bre Ligia, Zhao Tianyu, Zheng yu, Newland Ben, Wang Wenxin. Mussel- inspired hyperbranched poly(amino ester) polymer as strong wet tissue adhesive. Biomaterials. 2014; 35.
- Ishida Y, Sun ACF, Jikei M, Kakimoto M. Synthesis of hyperbranched aromatic polyamides starting from dendrons as AB(x) monomers: effect of monomer multiplicity on the degree of branching. Macromolecules. 2000; 33: 2832e8.
- Kord Forooshani P, Lee BP. Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci. Part A Polym. Chem. 2017; 55: 9-33.
- Qu J, Zhao X, Liang Y, Zhang T, Ma PX, Guo B. Antibacterial adhesive injectable hydrogels with rapid self-healing, extensibility and compressibility as wound dressing for joints skin wound healing. Biomaterials. 2018; 183: 185-199.
- He X, Liu L, Han H, Shi W, Yang W, Lu X. Bioinspired and Microgel- Tackified Adhesive Hydrogel with Rapid Self-Healing and High Stretchability. Macromolecules. 2019; 52: 72-80.
- Blacklow SO, Li J, Freedman BR, Zeidi M, Chen C, Mooney DF. Bioinspired mechanically active adhesive dressings to accelerate wound closure. Science Advances. 2019; 5: eaaw3963.
- Yi H, Lee S, Seong M, Kwak M, Jeong H. Bioinspired reversible hydrogel adhesives for wet and underwater surfaces. J. Mater. Chem. B. 2018; 6: 8064.
- Chen T, Chen Y, Rehman HU, Chen Z, Yang Z, Wang M, et al. Ultratough, Self-Healing, and Tissue-Adhesive Hydrogel for Wound Dressing. ACS Appl. Mater. Interfaces. 2018; 10: 33523-33531.
- Spencer P, Schaumburg H. A Review of Acrylamide Neurotoxicity Part I. Properties, Uses and Human Exposure. Canadian Journal of Neurological Sciences/Journal Canadien Des Sciences Neurologiques. 1974; 1: 143-150.
- Hogervorst JG, Schouten LJ, Konings EJ, Goldbohm RA, van den Brandt PA. A prospective study of dietary acrylamide intake and the risk of endometrial, ovarian, and breast cancer. Cancer Epidemiol. Prev. Biomark. 2007; 16: 2304-2313.