
Mini Review
Austin Chromatogr. 2014;1(2): 4.
HPLC Calibration Process Parameters in Terms of System Suitability Test
Anirbandeep Bose*
Acharya and BM Reddy College of Pharmacy, India
*Corresponding author: Anirbandeep Bose, Acharya and Bm Reddy college of Pharmacy, India
Received: September 01, 2014; Accepted: September 24, 2014; Published: October 07, 2014
Abstract
During the routine analysis of drug and analytes System Suitability Test (SST) is one of the most important and integral parts of HPLC method development and calibration. System Suitability Test (SST) is generally performed to evaluate the suitability and effectiveness of the entire chromatographic system not only prior to use but also during the time of analysis. Due to several reasons the performance and the capacity of the entire chromatographic system may abruptly or mildly change during their regular uses. This modification of the system can in turn affect the reliability of the entire HPLC analytical results. For this reasons different parameters which is related to the operation of the whole chromatographic system can be monitored and investigated to find out the integrity and reliability of the whole HPLC systems. These parameters are used to determine characteristic chromatographic parameters, such as the number of effective theoretical plates, resolution, asymmetry, detection limit and selectivity. After the checking of the parameters, the system is then only can be declared suitable if the responses are within permitted limits. The objective of our present review article is to emphasize the importance of the System Suitability Test (SST) for development and validation of the HPLC analysis. In addition, we also discuss about the measurement and calculation of different SST parameters in very simpler way.
Keywords: HPLC; LC-MS; Pharmacokinetics; Bioanalytical method development
Introduction
Chromatography, specifically liquid chromatography in the form of HPLC is used enormously in pharmaceutical drug analysis. In pharmaceutical analysis generally 4 steps is important for the successful validation of HPLC analysis. The first step and second step can be defined as Initial System Qualification followed by Periodic Calibration. The third step is Method Validation, where performance of the entire analytical procedure including sample preparation has been verified and justified. The final and last step is the determination of System Suitability Testing (SST), which significantly contribute to determine the suitability of chromatographic system on a day-to-day basis. In this review article, we emphasize on this final validation step and the main focus of our entire review article is to discuss how to perform SST and define the exact limits according to the latest regulatory guidelines [1-3].
What is SST?
SST is mainly applied to determine the column efficiency, resolution, and repeatability of a particular chromatographic system to verify its ability for a defined analysis. According to the United States Pharmacopeia and the International Conference on Harmonization (ICH), SST is an essential feature of all HPLC analytical procedures. Although USP and ICH are not the mandatory part of regulatory agencies, but their guidelines are the integral parts of the industry because of its acceptance by the FDA. SST is based on the concept that the equipment, electronics, analytical operations, and samples to be analyzed constitute an integral system that can be evaluated as a whole. The chromatographic systems used for most pharmaceutical analyses such as assays of the active ingredients, impurity determinations, and dissolution testing (measuring the dissolution rate for a particular form of dosage) must pass a set of predefined acceptance criteria (SST limits) before sample analysis can commence [1-5].
Why SST is necessary?
USP and ICH guidelines have specially emphasize on the use of SST to satisfy the latest regulatory requirements. One of the most important facts about the SST is that it should be performed not only before the experiment but also at different time interval throughout the all HPLC assays. The reason behind the SST analysis at different time interval is based on the concept that it should not be assumed that the system will behave properly throughout experiment. In addition to this, SST studies corresponding to compound of our interest is not enough to check system suitability because the system’s separation capacity is not investigated thoroughly. But, System Suitability Samples (SSSs) or resolution test mixtures of components of interest and expected impurities is required. For this reasons SSTs are analysed before and during testing.
The most important SST parameters which are investigated for different HPLC analysis is Resolution (R), repeatability (RSD— relative standard deviations—of peak response and retention time), column efficiency (N), and Tailing factor (T). These parameters are possessing para amount interest as they indicate system specificity, precision, and column stability. Other parameters include capacity factor (k) and Signal-to-noise ratio (S/N) for impurity peaks. All most all HPLC data systems are based on the software dta system which can calculate the measurement and report of these SST parameters. most “official” analytical methods has fixed the acceptance criteria or SST limits which may vary with different tests and are typically less stringent for biologics and trace impurities [1-7].
Setting limits
SST limits should emphasize the minimum criterion rather than optimal values. Analytical methods simply adopted the general limits from the CDER guidance document [2]. Different guidelines related to SST limits are shown (Table 1)
![]()
SST limits
CDER guidelines
Hsu and Chien recommendation
Repeatability of peak response
≤1.0% for 5
≤1.5% general
5–15% trace
<5% biologicsResolution
>2.0 general
>2.0 general
>1.5 quantizationTailing factor
≤2.0
<1.5–2.0
Column efficiency
>2000 (plate count)
Not available
Capacity factor
>2
2–8
Table 1: SST limits for HPLC chromatogram according to different guidelines.
System Suitability Parameters
The system suitability parameters which are generally accepted by regulatory authorities and independent auditor are depicted below:
- Peak retention time,
- Peak area,
- Amount,
- Peak height,
- Peak width at half height,
- Peak symmetry,
- Peak tailing,
- Capacity factor (k´),
- Plate numbers,
- Resolution between peaks,
- Selectivity relative to preceding peak
Peak symmetry and tailing factor [8]
Our treatment of chromatography in this section assumes that a solute elutes as a symmetrical Gaussian peak, such as that shown in Figure 1 as dotted line. This ideal behaviour occurs when the solute’s partition coefficient, KD is the same for all concentrations of solute
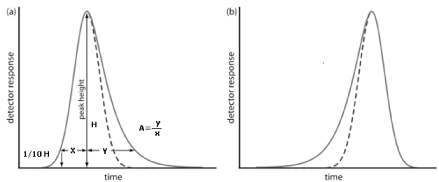
Figure 1: Examples of asymmetric chromatographic peaks showing (a) peak tailing and (b) peak fronting. For both (a) and (b) the un dotted chromatogram is the asymmetric peak and the dotted chromatogram shows the ideal, Gaussian peak shape.
KD = [Ss] / [Sm]
[S]s= concentration of solute in the stationary phase, and [S]m = concentration of in the mobile phase,
If this is not the same, then the chromatographic peak has an asymmetric peak shape similar to those shown in (Figure 1a). The chromatographic peak in Figure 1a is an example of peak tailing, which occurs when some sites on the stationary phase retain the solute more strongly than other sites. (Figure 1b), which is an example of peak fronting is most often the result of overloading the column with sample. As shown in (Figure 1a), we can report a peak’s asymmetry by drawing a horizontal line at 10% of the peak’s maximum height and measuring the distance from each side of the peak to a line drawn vertically through the peak’s maximum. The asymmetry factor, A, is defined as
A= y / x
The Tailing Factor is defined by the USP as the distance from the front edge of the peak to the back edge, divided by the distance from the front edge to the centreline, with all distances measured at 5% of the maximum peak height (Figure 2). Peak Tailing (T) can be expressed as T =(x+y)/2x

Figure 2: Calculation of peak tailing (T)=(x+y)/2x.
Capacity factor (k)
Theory of HPLC retention based on the adsorption from solutions can help to establish the relationships of measurable retention values (VR, tR, and k’) with the thermodynamic parameters, such as adsorption equilibrium constant (K), or free Gibbs energy (). Capacity factor is the ratio of the reduced retention volume to the dead volume:
The retention volume can be defined as the volume of mobile phase required to elute a substance from the chromatography column. Vr= Vm - KD Vs where Vm = void volume and KD = distribution coefficient and Vs = stationary phase volume.
Dead volume or void volume is the total volume of the liquid phase in the chromatographic column.
Void Volume can be calculated as the following equation
where:
r = radius of column [cm]
π= constant, ratio of circumference to diameter of a circle
L = length of column [cm]
f = fraction of column volume that is not taken up by stationary phase but available for mobile phase; default value for f = 0.7 (for Hypersil) [9]
Resolution
Selectivity and resolution [10]
Until now, retention and efficiency was discussed separately, but both of these parameters are affecting the separation of the mixture. Retention is developing the separation, and band broadening is destructing it. Selectivity is the ratio of the capacity factors of both peaks, or the ratio of its adjusted retention times. Selectivity represents the separation power of particular adsorbent to the mixture of these particular components (Figure 3).

Figure 3: Measurement of selectivity and resolution values measured from a chromatogram containing two peaks.
This parameter is independent of the column efficiency; it only depends on the nature of the components, eluent type, eluent composition, and adsorbent surface chemistry. In general, if the selectivity of two components is equal to 1, then there is no way to separate them by improving the column efficiency. Resolution is the parameter describing the separation power of the complete chromatographic system relative to the particular components of the mixture. By convention, resolution (R) is expressed as the ratio of the distance between two peak maxima to the mean value of the peak width at the base line:
If we approximate peaks by symmetric triangles, then, if R is equal to or more than 1 then components are completely separated. If R is less than 1, then components are overlapped. By using the expressions for capacity factor and column efficiency the equation for R could be transferred to the form:
where the dependence of resolution on the column efficiency is represented by the square root of N, which means that increasing the efficiency is not so favourable for resolution improvement.
Resolution can also be expressed in form:
Theoretical Plate Count [11]
Plate count is a (theoretical) measure of the efficiency of the HPLC column. In HPLC chromatography there’s equilibrium between the stationary phase and the mobile phase. During the time of elution there’s a transfer of molecules from the mobile phase to the stationary phase and back to the mobile phase, and so on back and forth down the length of the column--that’s how the separation occurs. The distance along the column that it takes to make one of these transfers between phases and re-equilibrate is called the theoretical plate Height (H). If it takes less distance, that means the plates are narrow and you have more of them in the column, which means more transfers between phases can take place, and the column is more efficient. That of course means better resolution and better peak separation. If it takes a comparatively greater distance for one phase transfer event, the plates are wider and there are fewer of them, so your efficiency (and therefore your resolution) is not as good. The relationship between theoretical plate height, plate count, and column length is theoretical plate Height (H) x plate count (N) = column Length (L) or plate count = column length/ theoretical plate height Remember, this is only theoretical, so round off any calculations you do for plate count. Hope this helps you. N, the number of theoretical plates, is one index used to determine the performance and effectiveness of columns, and is calculated using equation.
Where tr: retention time, and W: peak width
This peak width, W, is based on the baseline intercepts of tangent lines to a Gaussian peak, which is equivalent to the peak width at 13.4 % of the peak height. However, to simplify the calculation and accommodate non-Gaussian peaks, the following calculation methods are used in actual practice.
References
- Dong M, Paul R, Gershanov L. Getting the peaks perfect: System suitability for HPLC. Today’s Chemist at Work.
- Center for Drug Evaluation and Research, U.S. Food and Drug Administration. Reviewer Guidance, Validation of Chromatographic Methods; FDA, Rockville. 1994.
- Mukherjee A, Bera A. A Detailed Study of Validation Parameters and System Suitability Test in HPLC. Res J Pharm Bio Chem Sci. 2012; 3: 426-447.
- Hsu H, Chien CS. Validation of analytical methods: simple methods for HPLC assay method. Food Drug Anal. 1994; 2: 161-176.
- Wiggins DE. System Suitability in an Optimized HPLC System. J Liq Chromatogr. 1991; 14: 3045-3060.
- Hokanson GC. A life cycle approach to the validation of analytical methods during pharmaceutical product development, Part I: The initial validation process. Pharm Tech. 1994; 118–130.
- Furman WB, Dorsey JG, Snyder LR. System Suitability Tests in Regulatory Liquid and Gas Chromatographic Methods: Adjustments Versus Modifications. Pharm Technol. 1998; 22: 58-64.
- General Theory of Column Chromatography.
- Tailing and Symmetry. Chromatography Forum.
- Selectivity and Resolution.
- What does "plate count" mean in HPLC?