
Research Article
Austin Chromatogr. 2014;1(3): 5.
Quantification of Polynuclear Aromatic Hydrocarbons in Retention Pond Waters Using Gas Chromatography-Tandem Mass Spectrometry
Poulain M1, Geffroy Rodier C1*, Canaff C1, Ruban V2 and Ambles A1
1Institute of Chemistry of Poitiers : Materials and Natural Resources, University of Poitiers, France
2Ifsttar Nantes, French Institute of Science and Technology, France
*Corresponding author: Geffroy Rodier C, Institute of Chemistry of Poitiers : Materials and Natural Resources, UMR 7285, B 27, 4 rue Michel Brunet, B27-TSA51106, 86073 Poitiers, France
Received: September 25, 2014; Accepted: October 21, 2014; Published: October 30, 2014
Abstract
The individual quantification of 16 Polynuclear Aromatic Hydrocarbons (PAHs) in French retention pond water samples is reported. The full analytical protocol including Solid Phase Extraction (SPE) and tandem mass spectrometry detection allowed the detection limit in the range of 0.5–5 ng L-1. PAHs quantification is achieved using a seven point calibration plot with internal calibration. Real sample analysis proved that the procedure is convenient for PAHs determination in environmental water. Due to the high levels of PAHs in retention pond waters care must be taken when considering the reuse of such samples.
Keywords: Triple quadrupole mass spectrometry; PAHs; Water; Retention ponds
Introduction
During the last decades, tremendous improvements in analytical instrumentation have allowed a better detection of pollutant in diluted systems such as water. However monitoring of pollution of ecosystems is still an ongoing challenge. Due to their mutagenic and carcinogenic properties [1,2], PAHs are included in the US Environmental Protection Agency (EPA) priority pollutant list [3]. PAHs are derived from natural or anthropogenic sources. Natural sources include forest and prairie fires [4] or post-depositional transformations of biogenic precursors [5]. Anthropogenic sources include combustion of fossil-fuel [6], long-range atmospheric transport of PAHs [7,8]; urban runoff [5] and spillage of petroleum and its refined products [9]. These organic pollutants are ubiquitous. They are found, for example, in food [10], water [11], soils, sediments [12] and air particulates [13]. In recent years, they have received considerable attention as, in addition to their toxicity, they are highly persistent and can accumulate in environmental aqueous systems. Presence of PAHs in water is now part of environmental concern.
Over the past few decades, retention ponds have been built alongside highways and motorways with the initial role of controlling water flow during rainstorms. Urban and road runoff waters contribute to pollution of receiving ponds, streams and lakes. Previous studies show that PAHs are highly concentrated in sludge of some retention ponds, i.e. 400-900 times the Dutch threshold value for polluted soils [14,15]. Thus, concentrations of such compounds in water retention ponds must be evaluated.
Many analytical techniques have been developed for the determination of organic compounds in water samples. PAHs can be extracted from aqueous matrixes by various methods such as liquid/ liquid extraction [11-16], Solid Phase Extraction (SPE) [17,18], Solid- Phase Micro Extraction (SPME) [19,20] or Stir Bar Sorptive Extraction (SBSE) [21,22]. Detection is performed by Gas Chromatography (GC) or High Performance Liquid Chromatography (HPLC). Detection limits for the EPA–PAHs depend on the selected concentration method and the detection. SPE coupled to HPLC / fluorescence is the most common procedure to quantify PAH traces in water with a detection limit in the range of ngL-1. Compared to single stage MS modes, tandem Mass Spectrometry (MS–MS) offers a higher degree of sensitivity. MS–MS enables the analysis of organic molecule trace levels in the presence of interfering compounds without losing identification capability due to a drastic reduction of the background signal [23,24]. Triple quad technology directly enables the recovery of three different mass spectra (precursor ions, product ions and neutral fragments).
This paper presents GC-MS-MS as a sensitive alternative to HPLC for the analyses of PAHs in environmental samples. Validation parameters such as linearity, reproducibility, limits of detection and quantification are determined. Finally, the procedure is applied to the quantification of PAHs from two French retention pond waters.
Material and Methods
Chemicals and reagents
HPLC grade solvents (acetonitrile, dichloromethane and methanol) are purchased from Merck (Darmstadt, Germany) and are re-distilled before use. Water is purified on a Milli-Q SP reagent water system (Millipore, Bedford, MA, USA). Standard mixture of the 16 priority PAHs, at 10 mgL-1 in acetonitrile, is purchased from Sigma- Aldrich. The surrogate internal standard is a mixture containing perpetuated PAHs ([2H8] naphthalene (Np-d8), [2H10] acenaphthene (Ace-d10), [2H10] phenanthrene (Ph-d10), [2H12] chrysene (Chry-d12), [2H12] perylene (Pe-d12) purchased from Sigma-Aldrich.
SPE is performed onto Waters Oasis HLB Sorbent cartridges (6cc/200mg) purchased from Waters. Stir bars (Twisters), 10 mm long, coated with a 0.5 mm film thickness layer (24 L) of PDMS, are from Gerstel (Mulheim, Germany).
Sample description
The Wissous pond was created in 1999. Located in the industrial area of Villemilan (France), it drains the A6 motorway area. The Saint Joseph pond is located at the North of Nantes (on the French West coast). It was created in the midth of the 20th century; it is of urban type, draining the St Joseph district.
Solid phase extraction
SPE cartridges are conditioned with 5mL dichloromethane and 5mL methanol. Before loading on cartridges acetonitrile is added to avoid PAHs adsorption upon glassware. Standard solutions and water samples (50mL) are loaded at a flow rate of 1 mL.min-1. Then the cartridge is rinsed with 5 mL Milli-Q water. Organics are extracted using 8 mL dichloromethane, evaporated and reconstituted in 200 μL dichloromethane. Pure dichloromethane processed through SPE units is used as the procedural blank.
Stir bar sorptive extraction
Stir bars extraction is performed according to Garcia Falcon et al. (2004) on PDMS coated bars. PAHs desorption is performed with an ultrasonic device (Sonorex Digital 10P, Bandelin GmbH, Germany) for 10 minutes in 200 μL acetonitrile.
Gas chromatography-mass mass spectrometry
GC-MS-MS analyses are carried out on a Varian 3800 GC gas chromatograph coupled to a Varian 1200 L triple Quadrupole mass spectrometer (Varian, Les Ulis, France). The GC system is equipped with a PTV injector with a SGE Liner. All analysis is carried out in split less mode at the temperature of 300 °C. The split less valve is closed for 0.75 min, and then operated in the split mode at 20 mL/ min. All injections volumes are 1μL.
Separation is achieved on a 30 m x 0.25 mm i.d.VF-5ms (Factor Four, Varian, France) coated with 5% phenyl, 95 % dimethylpolysiloxane (film thickness 0.25μm). The temperature program is 1 min at 50 °C, 10°C/min up to 100 °C, 9°C/min up to 310 °C and temperature is finally hold for 4 minutes at 310 °C. The carrier gas is Helium and the column head pressure is 15 psi to reach a constant flow rate of 1.2 ml/min.
The Quadrupole mass spectrometer is operated under electron impact ionization (70eV). The filament emission current is 500μA. The source and transfer line temperatures are at 250°C and 310°C, respectively. The electron multiplier is set at 1880 V. The acquisition mode chosen for mass spectrometry analysis is MRM mode (Multiple Reaction Monitoring). The scantime is 0.3 s/scan. Peak detection and integration are carried out using Varian Workstation version 6.3. Precursor ions are isolated using 0.7 amu isolation window and subjected to Collision Induced Dissociation (CID) in the second Quadrupole (Q2). The dissociation is induced by collisions with neutral target species of the collision gas: Argon. In the third step, ions are transmitted in the third Quadripole (Q3) for separation. In MRM mode the fragment ions are also selected in the third quadripole. Different collision energies and multiple transitions are studied for each compound.
PAHs quantization is achieved using a seven point calibration plot with internal calibration over the linearity GCMS-MS range (1- 100 μg/L) established with 40 μg/L internal perpetuated standards and 1,5,10,25, 50, 75 and 100μg/L PAH solutions. Limit of Detection (LOD) and Limit of Quantification (LOQ) are estimated at the lowest concentration of analytes having clear discerned peaks with Signal to Noise ratio (S/N) of 3 and 10 respectively.
Results and Discussion
Mass spectrometry and quantification
In order to improve sensitivity and selectivity, the MRM acquisition mode is used. In a triple quadripole instrument, the ions generated in the EI source are transmitted to the first Quadripole (Q1) where precursor ions are selected. A preliminary study is carried out in MS-MS mode under full scan conditions on Q3 in order to determine the fragment ions to be used for further MRM experiments. For each PAH from one to three ions are observed. In most cases these ions are molecular ions (M)+, (M-H)+, (M-H2)+ or (M-C2H2)+ fragment ions. The collision energy is selected for each PAH to obtain the maximum signal dissociation of the product ions (table 1). The optimization of the CID voltage of each transition is performed using multiple scan function in different channel for each retention time window (segment). The influence of CID voltage is studied from 10V to 40 V at 5V steps. The example for chrysene is given in Figure 1: 10eV for (M-H)+ (227), 35eV for (M-H2)+ (226) and 20 eV for (M-C2H2)+ (202). The argon collision gas pressure is set at 2.2 mtorr. The higher pressure of 4 mtorr is discarded because of the lower sensibility obtained at each transition (Figure 1).
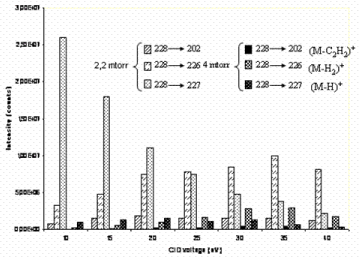
Figure 1: Influence of collision gas pressure on the intensity of the three chrysene’s transitions at different CID voltage.
LOD and LOQ of GCMSMS analysis are in the range of HPLC/ Fluorescence limits and are better than those obtained with ion trap tandem mass spectrometry [25].
![]()
Segment
time (min)
Compound
Retention time (min)
Precursor ion (m/z)
Channel
Product ion
CID voltage
(eV)
0-12
Naphthalene
9.62
128
3
2
1
127
102
78
10
20
20
12-15
Acenaphthylene
13.63
152
1
151
10
12-15
Acenaphthene
14.09
154
2
153
10
15-16
Fluorene
15.45
166
1
165
10
16-20
Phenanthrene
17.96
178
2
1
177
152
10
20
16-20
Anthracene
18.10
178
2
1
177
152
10
20
20-24
Fluoranthene
21.11
202
202
2
1
201
200
10
35
20-24
Pyrene
21.70
202
2
1
201
200
10
35
24-26.50
Benz[a]anthracene
24.89
228
3
2
1
227
226
202
10
35
35
24-26.50
Chrysene
24.99
228
3
2
1
227
226
202
10
35
20
26.50-30
Benzo[b]fluoranthene
27.55
252
2
1
250
226
35
35
26.50-30
Benzo[k]fluoranthene
27.61
252
2
1
250
226
35
35
26.50-30
Benzo[a]pyrene
28.29
252
2
1
250
226
35
35
30-33
Indeno[1,2,3-cd]pyrene
30.79
276
1
275
10
30-33
Dibenzo[a,h]anthracene
30.85
278
4
3
277
276
35
10
30-33
Benzo[ghi]perylene
31.47
276
1
275
10
Table 1: GC-MS-MS segment program with fragment ions chosen for each PAH.
SPE and SBSE extractions
To study environmental samples from urban and motorway retention ponds we first evaluate the extraction technique on a wide concentration range. SPE and SBSE are compared by spiking 50 mL Milli-Q water samples with the standard PAH solution in order to study concentrations ranging from 5 to 100 ng/L. Stir Bar Sorptive Extraction is a technique which has been introduced by Baltusen et al. [21], using stir bars (magnetic stirring rod incorporated in a glass jacket) coated with polydimethylsiloxane (PDMS). This phase is previously used for Solid Phase Micro Extraction (SPME) but due to a larger amount of PDMS relative to the SPME fiber, SBSE enables to increase recovery of analytes. Temperatures (room temperature, 40°C, 60°C, 80°C and 100°C) and adsorption time (from 1 to 10 hours) are studied (Figure 2). Compared to SPE, SBSE avoids the evaporation step which could explain the better recoveries (40%) for the lower molecular weight compounds at the lower concentration. For 5 ngL-1 SPE does not allow a recovery higher than 15% whereas 45% are recovered at 100 ngL-1. However the best conditions achieved after optimization are time and energy consuming steps (60°C and 6 hours). For the high molecular weight compounds SBSE recoveries are similar to those previously described in literature (from 40 to 50% with RSD (n=9) <10%) [22]. But are far lower than those achieved in present SPE experiments (average recovery by SPE 90%, Table 2). Regarding the diagnostic values of high molecular weight compounds we choose the SPE concentration method.
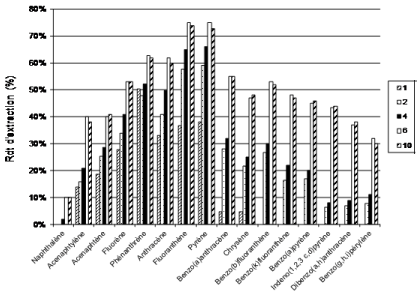
Figure 2: Influence of exposure time (from 1 to 10 hours) on SBSE recoveries at 60°C mesured at 25 ngL-1.
![]()
Compound
SPE
SBSE
Detection limit including SPE
Quantitation limit including SPE
Calibration curves y=ax+b
R2
Recovery* %
Recovery* %
(ngL-1)
(ngL-1)
Naphthalene
30
40
1.40
3.50
10.3558x
0,997
Acenaphthylene
30
40
1.40
6.60
4.0393x
0,999
Acenapthene
30
40
1.40
6.60
2.7364x+0.0688
0,995
Fluorene
27
53
1.40
3.70
0.8649x
0,998
Phenanthrene
72
63
1.50
1.50
6.3735x+0.3451
0,999
Anthracene
75
62
0.50
5.50
4.7588x+0.4
0,999
Fluoranthene
82
75
1.40
2.50
1.9916x+0.32
0,972
Pyrene
85
75
0.50
2.50
2.2094x
0,973
Benzo[a]anthracene
94
55
1.00
4.20
1.1434x
0,993
Chrysene
95
47
1.00
4.20
1.3334x
0,996
Benzo[b]fluoranthene
91
53
3.00
5.50
2.3022x
0,983
Benzo[k]fluoranthene
94
48
3.00
5.50
2.6742x
0,997
Benzo[a]pyrene
91
45
4.00
6.60
2.4684x
0,995
Indeno[1,2,3-cd]pyrene
90
44
3.00
5.80
4.5376x
0,996
Dibenzo[a,h]anthracene
94
37
4.00
6.40
1.3937x
0,987
Benzo[ghi]perylene
91
32
2.00
4.40
7.3390x
0,998
Table 2: Validation parameters of the SPE-GC-MS-MS method.
For the study of natural samples, the SPE concentration procedure thus is performed. For natural waters, including SPE concentration step, LOD ranges between 0.50 and 4.00 ng/L (Table 2) which is ten times better than SPE GC FID validated method for similar water samples [26]. All standard solutions are injected 5 times to estimate intra-day (RDS<4%) and inter-day (RSD <6%) precisions of the SPE-GC-MS-MS method (Table 2).
Analysis of samples from water retention ponds
In water retention ponds, PAHs may be present as very complex mixture with many isomers. The use of a sensitive and selective method is then needed. The Wissous and St Joseph water samples are analyzed by the present SPE-GC-MS-MS method. Results are presented in Table 3 and illustrated on Figure 3. Low molecular weight PAHs are not detected in the two studied samples. Overall PAH concentration (sum of the 16 EPA-HAPs) is 3 times higher in Wissous sample than in Saint Joseph one (0.31 and 0.98 μg/L respectively). Ratios listed in table 3 indicate a pyrogenic origin for Saint Joseph PAHs whereas some petroleum origin is inferred for the Wissous ones [9]. Thus, chrysene (Chry) and benz [a] anthracene (B[a] A), often looked upon as the most toxic of the PAHs, are present in both samples. Regarding the value of the ratio Bz[a]A / (Bz[a]A + Chry) the dominant source of these pyrogenic PAHs is combustion (burning of wood or coal, or natural forest fires) (0.79>0.5) in St Jospeh sample but is more debatable in Wissous one (0.56) [27]. The traffic pollution is however confirmed by the B[a]Pyr / B[ghi]Pe ratio close to the 0.3-0.44 range, characteristic of traffic emission [27]. This pollution is already described in Wissous sludge [14]. Part of the PAHs found in Wissous water samples could come from the two motorways located in the neighborhood. Indeed, in such retention ponds, urban run-off could contain PAHs from gasoline and oil drips or spills exhaust products, tyre particles, and bitumen’s from road surfaces [5]. This result implies that heavy metal pollution linked to traffic could also be assumed. Furthermore, high concentration of B[a]Pyr (strongly carcinogen) in Wissous could rise serious ecological and human health risks.
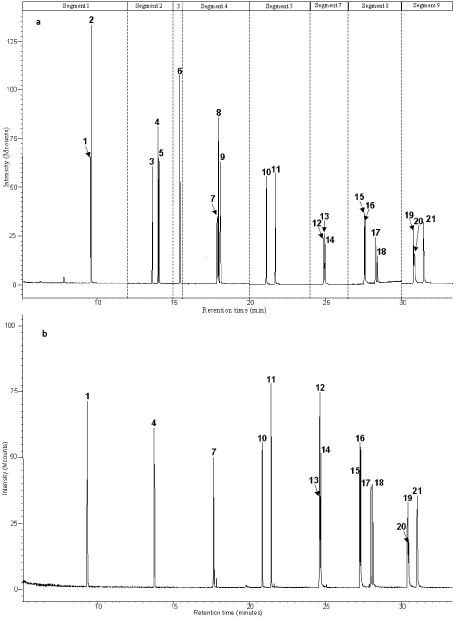
Figure 3: Chromatogram of the standards SEPARATIONand b Chromatogram of PAH from water of Wissou sample. Time segment program illustration. 1 Np-d8, 2 Np, 3 Acy, 4 Ace-d10, 5 Ace, 6 F, 7 Ph-d10, 8 Ph, 9 An, 10 Fl, 11 Py, 12 B[a]An, 13 Chry-d12, 14 Chry, 15 B[b]Fl, 16 B[k]Fl, 17 B[a]Py, 18 Pe-d12, 19 In, 20 Db[a,h]An and 21 B[g,h,i]Pe.
![]()
Saint Joseph
Wissous
Compound
ngL-1
ngL-1
Naphtalene
*ND
ND
Acenaphtylene
ND
ND
Acenaphtene
ND
ND
Fluorene
58
60
Phenanthrene
12
ND
Anthracene
ND
ND
Fluoranthene
14
18
Pyrene
ND
ND
Benzo(a)anthracene
22
94
Chrysene
6
74
Benzo(b)fluoranthene
22
110
Benzo(k)fluoranthene
28
130
Benzo(a)pyrene
22
96
Indeno(1,2,3 c,d)pyrene
64
134
Dibenzo(a,h)anthracene
ND
104
Benzo(g,h,i)perylene
66
158
Σ 16 PAH
314
978
Bz[a]A / (Bz[a]A + Chry)
0.79
0.56
B[a]Pyr / B[ghi]Pery
0.33
0.61
ΣB[b]Fl, B[k]Fl,� In and B[g,h,i]Pery
0.18
0.53
Table 3: PAHs distribution of Saint Joseph and Wissous water samples.
As streams and rivers, ponds are frequently used for potable water supply, contamination of watercourses, where water re-use is practiced, is particularly undesirable. European community directive, dealing with drinking water, has stated a maximum level for benz[a] pyrene (B[a]Py, strongly carcinogen) at 10 ng/L. In addition to be classified as drinking water, the sum of benz[b]fluoranthene (B[b] Fl), benz[k]fluoranthene (B[k]Fl), benzo[g,h,i] perylene (B[g,h,i]Pe) and indeno[1,2,3-cd] pyrene (In), must not reach 100 ngL-1 [28]. For Wissous and Saint Joseph the threshold values are by far exceeded. Thus, care must be taken if waters of the two samples have to be re-used.
Conclusion
As PAHs are persistent organic pollutants with highly toxic properties, monitoring their concentration in water samples is important for environment and of course for human health care. So, to analyze PAHs in such media a very selective and sensitive method has to be developed. Usually done by HPLC, in this paper, quantification is performed by gas chromatography coupled with tandem mass spectrometry. Before analysis water samples need to be concentrated by SPE which is the most efficient phase extraction to recover PAHs in the present working conditions. The GC-MS-MS methods enable to obtain low detection and quantization limits due to the ability to follow exclusively parents and fragments ions. The global method is successfully applied to water from retention ponds. The two studied samples exhibit high concentrations of natural and anthropogenic PAHs.
Acknowledgment
The authors would like to express their gratitude to J.Y. Viau (Saint Dizier Environment, France) for providing the water retention pond samples.
References
- Okona-Mensah KB, Battershill J, Boobis A, Fielder R. An approach to investigating the importance of high potency polycyclic aromatic hydrocarbons (PAHs) in the induction of lung cancer by air pollution. Food Chem Toxicol. 2005; 43: 1103-1116.
- Pufulete M, Battershill J, Boobis A, Fielder R. Approaches to carcinogenic risk assessment for polycyclic aromatic hydrocarbons: a UK perspective. Regul Toxicol Pharmacol. 2004; 40: 54-66.
- US Environmental Protection Agency, Guidelines establishing test procedures for the analysis of pollutants. Proposed regulations. Federal Register, USEPA, Washington, DC.
- Blumer M, Youngblood WW. Polycyclic aromatic hydrocarbons in soils and recent sediments. Science. 1975; 188: 53-55.
- Wakeham SG, Schaffner C, Giger W. Polycyclic aromatic hydrocarbons in Recent lake sediment. Geochimica Cosmochimica Acta. 1980; 44: 403-413.
- Hites RA, Laflamme RE, Farrington JW. Sedimentary polycyclic aromatic hydrocarbons: the historical record. Science. 1977; 198: 829-831.
- Lunde G, Bjorseth A. Polycyclic aromatic hydrocarbons in long-range transported aerosols. Nature. 1977; 268: 518-519.
- Laflamme RE, Hites RA. The global distribution of polycyclic aromatic hydrocarbons in recent sediments. Geochimica Cosmochimica Acta. 1978; 42: 289–303.
- Yunker MB, Macdonald RW, Vingarzan R, Mitchell RH, Goyette D, Sylvestre S. PAHs in the Fraser River basin: a critical appraisal of PAH ratios as indicators of PAH source and composition. Organic Geochemistry. 2002; 3: 489-515.
- Pincemaille J, Schummer C, Heinen E, Moris G. Determination of polycyclic aromatic hydrocarbons in smoked and non-smoked black teas and tea infusions. Food Chem. 2014; 145: 807-813.
- Tobiszewski M, Bigus P, Namiesnik J. Determination of parent and methylated polycyclic aromatic hydrocarbons in water samples by dispersive liquid-liquid microextraction-two dimensional gas chromatography-time-of-flight mass spectrometry. Analytical methods. 2014; 6: 6678-6687.
- Duong HT, Kadokami, Pan S, Matsuura N, Nguyen TQ. Screening and analysis of 940 organic micro-pollutants in river sediments in Vietnam using an automated identification and quantification database system for GC-MS. Chemosphere. 2014; 107: 462-472.
- Wu Y, Yang L, Zheng X, Zhang S, Song S, Li J, et al. Characterization and source apportionment of particulate PAHs in the roadside environment in Beijing. Sci Total Environ. 2014; 470-471: 76-83.
- Durand C, Ruban V, Ambles A, Oudot J. Characterization of the organic matter of sludge: determination of lipids, hydrocarbons and PAHs from road retention/infiltration ponds in France. Environ Pollut. 2004; 132: 375-384.
- El-Mufleh A, Bechet B, Grasset L, Rodier C, Gaudin A, Ruban. Distribution of PAH residues in humic and mineral fractions of sediments from stormwater infiltration basin. Journal of Soils and Sediment. 2013; 13: 531-542.
- Manoli E, Samara C. Polycyclic aromatic hydrocarbons in waste waters and sewage sludge: Extraction and clean-up for HPLC analysis with fluorescence detection. Chromatographia. 1996; 43:135-142.
- Barranco A, Alonso-Salces RM, Bakkali A, Berrueta L, Gallo B, Vicente F, et al. Solid-phase clean-up in the liquid chromatographic determination of polycyclic aromatic hydrocarbons in edible oils. Journal of Chromatography A. 2003; 988: 33-40.
- Erger C, Balsaa P, Werres F, Schmidt T. Multi-component trace analysis of organic xenobiotics in surface water containing suspended particular matter by solid phase extraction/gas chromatography-mass spectrometry. Journal of Chromatography A. 2012; 1249: 181-189.
- Eisert R, Levsen K. Development of a prototype system for quasi-continuous analysis of organic contaminants in surface or sewage water based on in-line coupling of solid-phase microextraction to gas chromatography Journal of Chromatography. 1996; 737: 59-65.
- Rupender K, Kumar P, Prashant C, Ansari N, Murthy R. Solid phase micro extraction combined with gas chromatography-mass spectrometry for the trace analysis of polycyclic aromatic hydrocarbons in chocolate. Analytical methods. 2013; 5: 1946-1954.
- Baltussen E, Sandra P, David F, Cramers C. Automated sorptive extraction-thermal desorption-gas chromatography-mass spectrometry analysis: Determination of phenols in water samples. Journal of Microcolumn Separation. 1999; 11: 737–747.
- García-Falcon MS, Cancho-Grande B, Simal-Gandara J. Stirring bar sorptive extraction in the determination of PAHs in drinking waters. Water Res. 2004; 38: 1679-1684.
- Schachterle S, Brittain RD, Mills JD. Analysis of pesticide residues in food using gas chromatography-tandem mass spectrometry with a benchtop ion trap mass spectrometer. Journal of Chromatography A. 1994; 683: 185-193.
- Pyle SM, Betowski LD, Marcus AB, Winnik W, Brittain RD. Analysis of polycyclic aromatic hydrocarbons by ion trap tandem mass spectrometry. Journal of the American Society for Mass Spectrometry. 1997; 8: 183-190.
- García-Falcon MS, Perez-Lamela C, Simal-Gandara J. Strategies for the extraction of free and bound polycyclic aromatic hydrocarbons in run-off waters rich in organic matter. Analytica Chimica Acta. 2004; 508: 177-183.
- Bispo J, Navickiene S, Dorea H. Method validation for SPE applied to determination of PAH in petroliferous industry effluent water. American Journal of Analytical Chemistry. 2011; 2: 971-978.
- Wu Y, Zhang J, Mi T, Li B. Occurrence of n-alkanes and polycyclic aromatic hydrocarbons in the core sediments of the Yellow Sea. Marine Chemistry. 2001; 76: 1-15.
- Official Journal of the European Communities (OJEC). Council Directive 98/83/EC (3rd November, 1998) relative to the quality of waters intended for human consumption. 1998.