
Research Article
Austin Chromatogr. 2014;1(4): 3.
Determination Polar Pesticides in Water Samples Using Acetonitril/Butylacetate Dispersive Solvent Extraction Combined with Gc-Ms Determination
Jalal Hassan1,2*, Maryam Ghafari3, Shahla Mozaffari3 and Abolfazl Farahani4
1Department of Toxicology, University of Tehran, Iran
2Department of Veterinary Medicine, University of Tehran, Iran
3Department of chemistry, Payame Noor University, Iran
4Department of Environmental, Iranian Mineral Processing Research Center, Iran
*Corresponding author: Jalal Hassan, Department of Toxicology, Faculty of Veterinary Medicine, University of Tehran, Iran
Received: October 18, 2014; Accepted: November 03, 2014; Published: November 04, 2014
Abstract
In this study, a simple, rapid and green sample preparation method has been developed for the extraction and preconcentration of polar pesticides in aqueous samples. In this proposed extraction procedure, an aqueous sample solution was added to a mixture of butyl acetate-acetonitril, was shake and was separated into two layers. The upper layer (butyl acetate) was collected and analyzed by GC/MS. No centrifugation was required in this procedure. Important parameters influencing the extraction process including type and volume of extraction solvent and co-solvent, and ionic strength were optimized. Under optimal conditions, the proposed method provided high extraction efficiency, good linearity range (0.7–50 ng mL-1), low limits of detection (0.005–0.2 ng mL-1) and good repeatability and recoveries. The relative standard deviations (RSDs, n = 5) were in the range of 2-9%. Finally, the proposed method was successfully applied to OPPs determination in water samples.
Keywords: Polar pesticides; Miniaturized liquid–liquid extraction; GC/MS; Preconcentration
Introduction
In general, sample preparation is the first step and possibly one of the most important steps in analysis. One of the trends in pesticide residue analysis is the development of rapid, sensitive, and accurate methodologies that can reliably identify and quantify the analytes in complicated matrices. Recent research in the analytical chemistry has focused on miniaturized, simplified, efficient and particularly environmental friendly extraction technique that inspires toward the development of micro extraction method. Consequently different micro extraction systems have been developed as solid-phase micro extraction [1], homogenous solvent extraction [2], and dispersive liquid-liquid micro extraction [3]. Low consumption of solvent, simple, rapid with high enrichment factor and recovery are the advantages of micro extraction method. Most of micro extraction applications are employed in aqueous samples for the extraction of nonpolar or moderately polar analytes. Dispersive Liquid–Liquid Micro Extraction (DLLME) is one of the emerging techniques in this area. Dispersive liquid–liquid micro extraction can be considered as a miniaturized version of conventional LLE and requires only micro liter volumes of solvents. DLLME is a new micro extraction technique with a high potential in sample pretreatment. It is based on a ternary component solvent extraction system (i.e. extraction solvent, disperser solvent and aqueous samples containing the analytes of interest). DLLME has extensively been used for direct extraction of pesticides from aqueous samples such water [4-6], but this technique has some limitation such as use the solvents with higher density than water (chlorinated solvents) bacause of its hazardous effects.
The aim of the present study is to develop the suitability of DLLME technique based on using low density organic solvent combined with GC/MS for the determination of polar pesticides (Table 1). The factors affecting the extraction efficiency were studied and the optimal conditions were used.
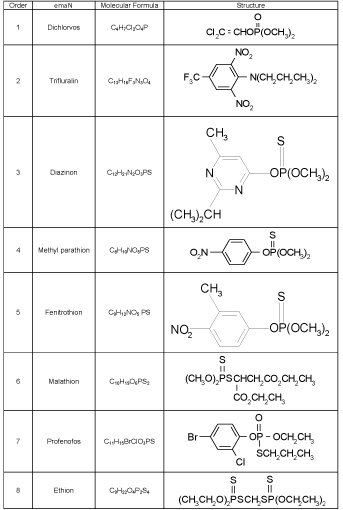
Table 1: Chemical structure of selected pesticide in this study.
Experimental
Chemicals and reagents
Analytical grade Methanol, n-hexane, cyclohexane, ethyl acetate, butyl acetate, hexyl acetate and NaCl were purchased from Merck (Darmstadt, Germany) and were used without further purification. Deionized water prepared on a Direct-Q 3 UV with a pump system (Millipore, Molshein, France). All pesticides (trifluralin, dichlorvos, diazinon, chlorpyrifos, methyl parathion, fenitrothion, malathion, profenofos and ethion) prepared from Ehrnestorfer (Augsburg, Germany). Each stock solution was prepared at a concentration of 100 mg L-1 in methanol and stored in a refrigerator (4 °C) until use. The working solution was prepared by appropriate dilution of the stock solution with the methanol.
Apparatus
The extracted compounds were analyzed on a 6890N Agilent gas chromatograph coupled to a 5975C Agilent mass-selective detector (Agilent Technologies, Avondale, PA, USA) and a 7683 Agilent autosampler. 2.0 μL of the sample were injected in the splitless mode at 250 °C into a 30 m×0.25mm×0.5μm DB-5 MS capillary column and operated by MSD Chemstation Software (Agilent Tecnologies). The mass spectrums were obtained at amass ratio scan range from 100 to 400 m/z to determine the appropriate masses for Selected Ion Monitoring (SIM). Helium (99.999% purity) was obtained from Roham Gas Company (Tehran, Iran) and was used as carrier gas at constant flow of 1.0 mL min-1.
Extraction procedure
0.5 mL of butyl acetate-methanol (1:2) was added to a dried volumetric flask (10 mL) and then, 9.5 mL of water sample was transferred to volumetric flask rapidly. The mixture was gently shaken for five minutes (Scheme 1). After this process, butyl acetate (extraction solvent) was separated at the top of volumetric flask. The butyl acetate was drawn out by a Hamilton syringe and transferred to a conical vial and 2.0 μL was injected to GC/MS.
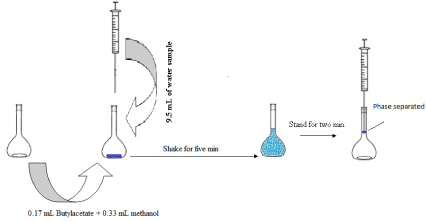
Scheme 1: Overall extraction step used in this method.
Results and Discussion
There are some factors affecting the extraction process, namely: kind of extraction and disperser solvents and volumes of both of them, and salt addition. The optimization of these parameters was carried out using 9.5 ml of an aqueous mixture standard solution containing a concentration of 10 ng mL-1 of each pesticide. The peak area was used to evaluate the extraction efficiency. The enrichment factor (Ef) was defined as the ratio between the analytes concentration in the organic phase () and the initial concentration of the analyte () in the aqueous sample:
and and the extraction recovery () is calculated using as follow:
Which and are the volumes of the extraction phase and aqueous sample, respectively.
Selection of the extraction solvent
The selection of an appropriate extraction solvent is very important for the dispersive solvent extraction process. It must be immiscible with water, providing the highest extraction efficiency, and have compatibility with analytical instrument for all the analytes. Solvents with lower density than water were chosen as follow: n-hexane, cyclohexane, and butyl acetate. A series of water samples spiking with pesticides were extracted with selected solvents, separately. These are explained by polarity of extracting solvent: the more polar solvent have higher ER than less polar solvent. The best extraction solvent appeared to be the butyl acetate (Figure 1).
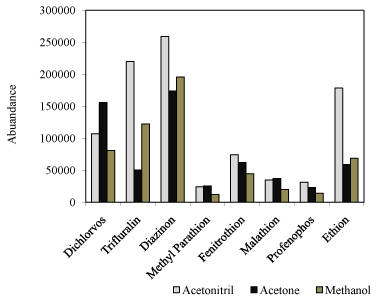
Figure 1: Effect of extracting solvent on extraction efficiency of pesticides. Conditions: volume of sample: 9.5 mL, concentration of pesticides: 10.0 ng mL-1, volume of solvent: 0.17 mL, volume of acetonitril: 0.33 mL.
Selection of the co-solvent
The co-solvent must be miscible with both sample solution and extraction solvent. Therefore, acetone, acetonitrile and methanol were tested as co-solvents, using 0.5 mL of each solvent containing 0.17 mL of butyl acetate (2:1). The best recoveries were obtained with acetonitrile except dichlroves (Figure 2).
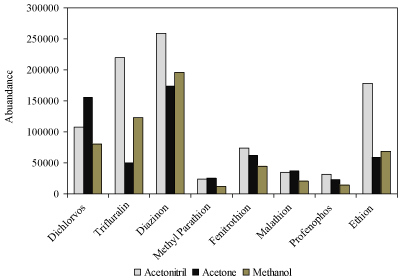
Figure 2: Effect of co-solvent on extraction efficiency of pesticides. Conditions: volume of sample: 9.5 mL, concentration of pesticides: 10.0 ng mL-1, volume of butylacetate: 0.17 mL, volume of co-solvent: 0.33 mL.
Volume of extracting solvent
In order to evaluate the effect of extraction solvent volume on extraction efficiency, butyl acetate solution volume was studied in the range of 0.17-0.67 mL and all the other parameters were maintained constant. The result shows that the overall enrichment factor decrease with increasing volumes of extraction solvent (data was not shown), because the volume of separated phase increases.
Salt addition
The effect of the salt addition on the extraction was evaluated with the sodium chloride concentration ranging from 0 to 2 % (w/v). The results show peak areas of OPPs were increased with increasing the NaCl concentration. Therefore, 2% of NaCl was selected for further studies.
Analytical performance
To plot calibration curves, pesticide standards in concentration of 1.0, 3.0, 5.0, 10.0, 25.0 and 50.0 ng mL-1 were prepared in of water sample, the solutions were extracted according to extraction procedure, and the peak areas were determined with triple injections. The calibration curve was generated by a least-squares linear regression analysis of the pesticides. As it can be seen, coefficient of regression (r) were higher than 0.993 for all cases. Table 2 shows the detection limits calculated as the lowest concentration of an analyte giving a signal of three-times the base line noise of the chromatogram and was in the range of 0.005-0.2 ng mL-1.
![]()
Pesticide
Retention time (min)
R2
DLR
(ng mL-1)
LOD
(ng mL-1)
Dichlorvos
6.3
0.9975
0.02-50.0
0.005
Trifluralin
15.8
0.9935
0.02-50.0
0.006
Diazinon
17.9
0.9977
0.08-50.0
0.025
Methyl parathion
18.1
0.9927
0.04-50.0
0.011
Fenitrothion
20.1
0.998
0.7-50.0
0.200
Malathion
21.4
0.9987
0.2-50.0
0.066
Profenofos
25.2
0.9985
0.3-50.0
0.08
Ethion
27.0
0.9957
0.03-50.0
0.007
Table 2: Figures of merit obtained for determination of pesticides after DLLME proposed method.
The accuracy of the proposed method was estimated using recovery experiments conducted at one concentration level. The repeatability, expressed as relative standard deviations (RSDs) for the three replicate analyses, was tested by spiking the water samples at a concentration level of 5.0 ng mL-1. A typical chromatogram obtained for spiked tap water is given in Figure 3. The RSDs (n = 3) varied between 2.0 - 6.6%. In order to investigate the developed method, the proposed method was applied to the analysis of pesticides in real water samples and the performance of the proposed method was investigated by determining the five pesticides in tap, well and river water samples (Table 3). No pesticide residues were found at the quantification level of the method.
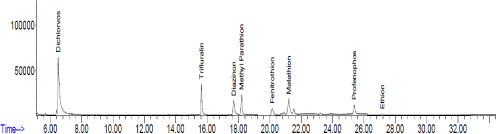
Figure 5: The chromatogram obtained for pesticide determination after DLLME method in tap water. Conditions: volume of sample: 9.5 mL, concentration of pesticides: 10.0 ng mL-1, volume of butylacetate: 0.17 mL, volume of co-solvent: 0.33 mL.
![]()
Sample
Average recovery � RSD% (n = 3 replicates)
Tap water
Well water
River water
Spiked level
(ng mL-1)
-
5.0
-
5.0
-
5.0
Dichlorvos
<LOD
95 �8
<LOD
93 �4
<LOD
94 �5
Trifluralin
<LOD
97 �5
<LOD
90 �4
<LOD
101 �8
Diazinon
<LOD
88 �7
<LOD
98 �5
<LOD
98 �9
Methyl parathion
<LOD
98 �2
<LOD
94 �9
<LOD
96 �4
Fenitrothion
<LOD
87 �6
<LOD
97 �7
<LOD
91 �6
Malathion
<LOD
100 �4
<LOD
105 �2
<LOD
93 �4
Profenofos
<LOD
99 �4
<LOD
92 �3
<LOD
98 �4
Ethion
<LOD
108 �3
<LOD
95 �5
<LOD
94 �5
Table 3: Recoveries and variations obtained for 8 pesticides artificially spiked in various water samples and analyzed with GC/MS.
Conclusion
The results show that, DLLME coupled with GC-MS offers a simple, rapid, green, and efficient technique for extraction and determination of pesticides in water samples. In this method, compared with other DLLME conventional methods, centrifugation was not needed and low density solvent, butyl acetate was used.
References
- Chunfeng D, Zheng S, Dapeng W, Yafeng G. Recent developments in solid-phase microextraction for on-site sampling and sample preparation. Trends in Analytical Chemistry. 2011; 30: 1568-1574.
- Hassan J, Farahani A, Shamsipur M, Damerchili F. Rapid and simple low density miniaturized homogeneous liquid-liquid extraction and gas chromatography/mass spectrometric determination of pesticide residues in sediment. J Hazard Mater. 2010; 184: 869-871.
- Andruch V, Kocurova L, Balogh LS, Skrlíkova J. Recent advances in coupling single-drop and dispersive liquid–liquid microextraction with UV–vis spectrophotometry and related detection techniques. Microchemical Journal. 2012; 102: 1-10.
- Caldas SS, Costa FP, Primel EG. Validation of method for determination of different classes of pesticides in aqueous samples by dispersive liquid–liquid microextraction with liquid chromatography–tandem mass spectrometric detection. Analytica Chimica Acta. 2010; 665; 55–62.
- Farajzadeh MA, Afshar Mogaddam MR, Abdollahi Aghdam A. Comparison of air-agitated liquid–liquid microextraction technique and conventional dispersive liquid–liquid micro-extraction for determination of triazole pesticides in aqueous samples by gas chromatography with flame ionization detection. Journal of Chromatography A. 2013; 1300: 70-78.
- Sanagi MM, Abbas HH, Ibrahim WAW, Aboul-Enien HY. Dispersive liquid–liquid microextraction method based on solidification of floating organic droplet for the determination of triazine herbicides in water and sugarcane samples. Food Chemistry. 2012; 133: 557–562.