
Research Article
Austin Chromatogr. 2015; 2(3): 1037.
Rapid Analysis of Moenomycin A Residue in Poultry Tissues Using Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectrometry
Xu H¹, Zhang HW¹*, Xu B¹, Zhang XM¹, Wang FM¹, Cao P², Yuan T³ and Wang YT¹
¹Technical Center of Inspection and Quarantine, Shandong Entry-Exit Inspection and Quarantine Bureau, People’s Republic of China
²Technical Center of Inspection and Quarantine, Yantai Entry-Exit Inspection and Quarantine Bureau, People’s Republic of China
³Shandong Entry-Exit Inspection and Quarantine Bureau, People’s Republic of China
*Corresponding author:Zhang Hongwei, Technical Center of Inspection and Quarantine, Shandong Entry- Exit Inspection and Quarantine Bureau, No.70 Qutangxia Road, Qingdao, Shangdong Province, P.R. China
Received: June 16, 2015; Accepted: September 24, 2015; Published: September 29, 2015
Abstract
A rapid, sensitive and accurate method has been developed for determination of Moenomycin A residue in poultry tissues. It was based on procedures with simple pretreatment process and Ultra High Performance Liquid Chromatography coupled to triple quadrupole tandem Mass Spectrometry (UHPLC-MS/MS). The procedures were deliberately optimized to tailor for complicated molecule and matrix. No significant matrix effects were observed after simple pretreatment and fast analysis within 10 min. Competent linearity across the concentration levels from 5 to 1000 μg.kg-1 was found with linear determination coefficient (r2) higher than 0.990. Limit of Detection (LOD) and Quantification (LOQ) were down to 0.25 μg.kg-1 and 0.8μg.kg-1 respectively. The mean recoveries from spiked blank kidney tissues at the levels of 10, 50, 500 μg.kg-1 ranged from 67.2 to 89.4%. Repeatability and within-laboratory reproducibility were lower than 7% and 11%. Finally, the proposed method was applied to real tissue samples and positive results were confirmed with the highest concentration up to 74.5 μg.kg-1.
Keywords: Moenomycin A; Residue; UHPLC-MS/MS; Poultry
Abbreviations
UHPLC-MS/MS: Ultrahigh Performance Liquid Chromatography Coupled to Tandem Mass Spectrometry; MRLs: Maximum Residue Limits; LOD: Limit Of Detection; LOQ: Limit Of Quantification; ESI: Electrospray Ionization; MRM: Multiple Reaction Monitoring; IDA: Information Dependent Acquisition; EPI: Enhanced Product Ion Scanning; CE: Collision Energy; SPE: Solid Phase Extraction; MEs: Matrix Effects; cmc: Critical Micelle Concentration
Introduction
Moenomycin, also named flavomycin, bambermycin, and flavophospholipol, is a mixture of structurally similar phosphoglycolipid antibiotics produced by various strains of Streptomyces species [1-3]. Moenomycin was first reported in the early 1960s and have been demonstrated to be effective against gram-positive bacteria via inhibiting the transglycosylation reaction, and thus interfering the synthesis of the bacterial cell wall [2,4]. Five major components of the moenomycin have been structurally characterized with the name of moenomycin A [5,6], moenomycin A12 [7], moenomycin C4, moenomycin C3 [8] and moenomycin C1 [9], of which moenomycin A is the main component. As shown in (Figure 1), this class compounds contain three general segments including a complex pentasaccharide, an unusual isoprenoid chain (C25, moenocinol) and a phosphoglycerate unit. Because of its high molecular weight and lipid tail in structure, moenomycin shows poor pharmacokinetics (i.e. long half-life in bloodstream and very low absorption from gastrointestinal tract) and has not been developed for human use but as an animal feed growth promoter for decades [10]. The mechanisms of growth promotion are still not exactly known. Several hypotheses have been proposed including nutrients protection against bacterial destruction, decline in toxin production from intestinal bacterial, and reduction in the incidence of subclinical intestinal infections [11]. Although studies in the past showed that oral dose of moenomycin at growth promotion level did not produce detectable residue in animal tissues, a slight absorption was detected when high doses were administrated [12-15]. Possibly retaining of moenomycin in animal tissues due to long time exposure or high dose administration could be of a concern. Therefore, according to “precautionary principle”, the European Commission had banned the use of the drug as an additive for feed stuffs since January 2006 [16] to avoid the possible risk of induced selection of resistant bacteria against moenomycin and related antibiotics (cross-resistance) that could be transmitted to human pathogens. Similarly, Maximum Residue Limits (MRLs) for flavophospholipol (moenomycin) in food products were specified in “Positive List” varying from 0.01 to 0.03 mg.kg-1 in different poultry tissues in Japan [17]. But in the US, China and many other countries, moenomycin is still approved for cattle, swine and poultry. For answering the concern and updating the data whether moenomycin could result in residue or not, there is a need to develop a method for analysis moenomycin residue in animal tissues with the current mainstream techniques.
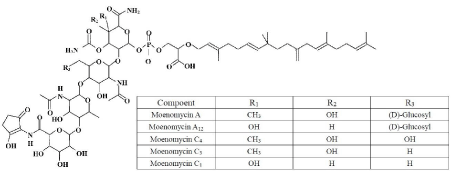
Figure 1: The molecular structure of moenomycin A and the other identified
moenomycin antibiotics belonging to the moenomycin family.
Compared with abundant publications about residue analysis of antibiotics, few of them were related to moenomycin. Among several recently reported methods, the matrices for moenomycin analysis were feed stuffs [18-20], and chicken litter [21]. To develop a rapid, sensitive and accurate method for determining moenomycin residue in poultry tissues which meet the requirements of regulations [17] and established criteria for banned drugs [22], the procedures based on a rapid sample preparation and UHPLC-MS/MS analysis were proposed in this work. Furthermore, characteristics of the method performance such as selectivity, linearity, recovery and repeatability, within laboratory reproducibility, Limit of Detection and Quantification (LOD and LOQ) were also investigated.
Materials and Methods
Chemicals and reagents
HPLC grade solvents (methanol, acetonitrile and hexane) for pretreatment and hyper grade acetonitrile for UHPLC-MS/MS analysis were all purchased from Merck (Darmstadt, Germany). Water was purified on a Milli-Q Advantage A10 apparatus (Millipore, Bedford, MA, USA). Formic acid and 25% ammonium hydroxide solution were supplied by Riedel de Haen (Seelze, Germany). Commercial standard of Moenomycin A was obtained from Sigma- Aldrich (Seelze, Germany) with the purity of 85.9%.
Stock standard solution of moenomycin A was prepared in methanol at 1000 mg.L-1. Intermediate standard solution at 10 mg.L-1 was prepared by dilution with methanol, which was stored in the dark at -18°C for at least 6 months. Working standard solution was prepared by diluting intermediate standard solution in methanol. It was stored at 4°C and prepared fresh weekly.
Sample preparation
A preliminary study was conducted using moenomycin A incurred broiler chickens. Tissues of muscle, fat, liver and kidney were analyzed for possible residue of moenomycin A. Results showed that the measurable amount of moenomycin A was found in kidney tissues, which was coincident with the literature [13]. Therefore, from the perspective of monitoring moenomycin A in animal tissues, kidney could be a suitable target matrix. The following study was preceded with kidney tissues.
About 2 g of homogenized tissue was weighed into 50 mL polypropylene centrifuge tube. 5 mL 25% ammonium hydroxide solution/methanol (1:9) was added. After vigorously shaking, the mixture was centrifuged at 12000 rpm for 10 min at 0°C. Supernatant was transferred into a new tube. After that, 30 mL acetonitrile was added and vortexes for 1 min. The tube was placed in a freezer at -20°C for 20 min and centrifuged as described above. Supernatant was abandoned. Another 5 mL 25% ammonium hydroxide solution/ methanol (1:9) was added into the tube. Then the tube was vortexes for 30 s and placed in an ultrasonic bath for 3 min at maximum power. Following, centrifugation was performed under the aforementioned conditions. Supernatant was transferred into a glass tube and evaporated to dryness under a stream of nitrogen at 45°C. Finally, the residue was reconstituted with 1 mL methanol and filtered through 0.22 μm PTFE syringe filter prior to LC-MS/MS analysis.
Ultrahigh performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS)
Analysis was carried out using Agilent 1290 Infinity ultra high performance liquid chromatography (Agilent Technologies, Boblingen, Germany) coupled to AB SCIEX QTRAP 5500 mass spectrometer (MDS SCIEX, Singapore). Chromatographic separation was achieved on a 100 mm×2.1mm i.d. 2.7 μm Agilent Poroshell 120 SB-C18 column (Agilent Technologies, Newport, USA) at 30°C with injection volume of 5 μL. The mobile phases, which comprised of a mixture of (A) 0.3% formic acid with 5% acetonitrile in water and (B) 0.3% formic acid with 5% water in acetonitrile, were delivered at 0.3 mL.min-1 under a gradient elution program: 0 min 50% A, 0.5 min 70% A, 5.0 min 5% A, 6.0 min 5% A, 6.1 min 50% A, 10 min 50% A. The mass spectrometer was operated in negative ESI mode, with an ion-spray voltage of -4.5 kV, source temperature of 500°C. The pressures of nebulizer, auxiliary and curtain gas (N2) were set to 40, 60 and 35 (arbitrary units) respectively. The Multiple Reaction Monitoring (MRM) transitions with related parameters used to detect Moenomycin A are summarized in Table 1.
![]()
Analyte
Retention time
MRM transitions
Declustering potential
Entrence potential
Collision energy
Collision cell
exist potential
(min)
(eV)
(eV)
(eV)
(eV)
Moenomycin A
4.4
789.9/575.9*
-155
-10
-42
-10
789.9/554.3
-37
-11
*Transition used for quantification.
Table 1: Summary of key ionization parameters for Moenomycin A, with chromatographic retention time.
For confirmation analysis, an advanced acquisition mode named MRM-IDA (information dependent acquisition)-EPI (enhanced product ion scanning) was used. In this mode, quantification transition was used as a survey scan, and a dependent scan, EPI, would be triggered when responses of survey scan surpassed 2000 cps (counts per second, threshold value set in IDA) after dynamic background subtraction. EPI scan rate was set at 10000 Da.s-1 with scan scope of 100 - 800 Da. Collision Energy (CE) for EPI was -35 (arbitrary unit) with spread CE set at 15 (arbitrary unit). Other settings were the same as MRM mode.
Performance characteristics of the method were evaluated including selectivity, linearity, trueness, precision, LOD and LOQ. The quantification of moenomycin A was conducted using matrixmatched calibration curve prepared by reconstituting residues from blank kidney tissue extracts with standard solutions. The acceptance criterion was that the determination coefficient (r2) must be more than 0.990. LOD and LOQ were estimated at the lowest concentration of analytes with signal to noise ratio (S/N) of 3 and 10 respectively.
Results and Discussion
Extraction of moenomycin A from kidney tissues
Moenomycin A exhibits limited solubility in solvents of low to medium polarity. However, better solubility in methanol was reported [18]. Methanol and alkalified methanol with 25% ammonium hydroxide solution have ever been employed to extract residue of moenomycin A in feed stuffs and chicken litter [20,21]. In this work, methanol, acetonitrile, acetone, were assessed independently or with several modifiers, such as formic acid, acetic acid and 25% ammonium hydroxide solution for extraction of moenomycin A by spiking-recovery tests. Pure solvents mixed with different percentage of modifiers were also evaluated. Results showed that 25% ammonium hydroxide solution/methanol (1:9) gave the highest recovery and therefore was chosen as the extraction solvent in the study. After extraction, clean-up measures should be taken to avoid severe Matrix Effects (MEs) which was witnessed by direct analysis of the extracts. Solid Phase Extraction (SPE), with different cartridges, is the most popular technique employed for further clean-up in residue analysis of animal products [23]. Rapid analysis, however, does not stand for time-consuming SPE procedures. Especially, the proposed SPE procedure in the literature [20,21] gave unsatisfactory results with recovery of ~30% when applied to kidney tissue samples. As an alternative, a faster technique than conventional SPE, dispersed SPE combined with variety of sorbents such as C18, diatomaceous earth and sea sand was applied to kidney samples in the work. Unfortunately, bad recoveries made the attempts abandoned due to occurrence of analytes adsorption to sorbents. Due to its complicated structure, Moenomycin A shows some special physicochemical properties such as aggregation in aqueous solution (with a cmc of 0.5 mM at pH 6.8), high affinity to proteins and cell membranes [24,25], which makes it difficult to extract and clean-up. To solve the problem, acetonitrile was used as a “reverse extraction” solvent to “solvent-out” the moenomycin A from the extracts by changing solubility of the analytes and utilizing high affinity of moenomycin A to proteins in extracts to form co-precipitation. Moenomycin A was then re-extracted by the extraction solvent again. The process effectively alleviates the MEs, evidencing by the results of MEs evaluation (discussed below). In the developed procedures amount of acetonitrile should be adequate to assure total solvent-out of moenomycin A at the concerned levels. To determine the amount of acetonitrile, aliquots of moenomycin A standard solution were spiked into blank kidney tissue samples at the level of 10, 100 μg.kg-1 and the samples were processed with proposed procedures except that amount of acetonitrile was set to 5, 10, 15, 20, 15, 30, and 40 mL respectively. At each level samples were analyzed in triplicates. As shown in (Figure 2), with the increase in the amount of acetonitrile, recovery of moenomycin A ascended and the curve reached a top flat when 30 mL of acetonitrile was used. At the same time, supernatant derived from acetonitrile at this level was dried under nitrogen gas and the residue was reconstituted in 1 mL methanol for LC-MS/MS analysis. Not any response was found at the time window of moenomycin A. Therefore, 30 mL acetonitrile was thought to be enough to “solvent-out” moenomycin A at the concerned levels, and the proposed procedures were also thought to be suitable for analysis of moenomycin A in kidney tissues.
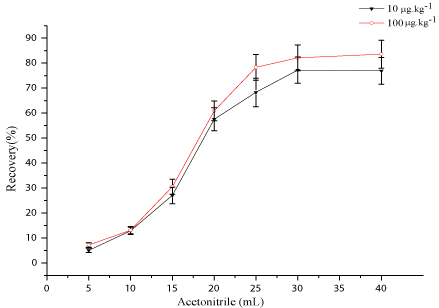
Figure 2: Optimization of the amount of acetonitrile added into the extract by
spiking-recovery tests at the two levels of 10 and 100 μg.μg.kg-1.
LC-MS/MS analysis
Several reversed-phase columns from different vendors were tested, and the final UHPLC separation was developed using an Agilent Poroshell 120 SB-C18 column (100mm×2.1mm, 2.7μm), that was packed with core-shell particles. This type of particles could offer better resolution, higher sensitivity and improved peak shape, especially for big molecules. The mobile phases were optimized in terms of sensitivity, peak shape and retention time by combining acetonitrile, methanol, water and several modifiers such as formic acid, acetic acid, ammonium format and ammonium acetate. It was found that acetonitrile/water with 0.3% formic acid showed superiority with described gradients.
Gallo et al. [20] have reported that the [M-2H] 2- at m/z 789.9 gave higher response than the [M-H] - at m/z 1580.4 in full mass scan under negative ESI mode. In our work, the double charged ion of 789.9 also showed better response than its single state, and therefore 789.9 was selected for parent ion of moenomycin A. To form transitions for monitoring moenomycin A, 575.9 and 554.3 were selected as productions for quantitative and qualitative purposes. The parameters for the transitions were optimized by flow injection analysis using moenomycin a standard solution at concentration of 200 μg.L-1 and the results were summarized in (Table 1).
Method performance
The selectivity was investigated by analyzing 20 blank kidney tissue samples in order to verify the absence/presence of interfering substances. The results demonstrated that no interfering peaks appeared within the 2.5% margin of the retention time of the analytes. The typical MRM chromatograms of the blank and spiked kidney tissue samples were presented in (Figure 3).
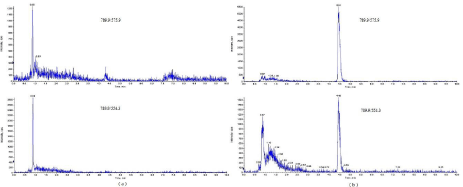
Figure 3: Extracted ion chromatographs for moenomycin A in (a) blank chicken kidney tissue and (b) spiked chicken kidney tissue at the level of 10 μg.μg.kg-1.
Standard solution calibration curve and matrix matched calibration curve were prepared simultaneously across the levels of 5, 10, 20, 50, 100, 200, 500, 1000 μg.kg-1 and at each level the tests were performed in triplicates. Matrix effects (MEs) were evaluated using slope ratio comparison according to the approach proposed by Hoff RB et al. [26]. The slope ratio of matrix-matched calibration curve to that of standard solution calibration curve below 0.9 or above 1.1 indicates ion suppression and ion enhancement, respectively. For value inside the range, MEs were considered negligible. As shown in (Table 2), the value of slop ratio was 1.14, indicating occurrence of a little matrix enhancement. To this end, the linearity was evaluated by matrix-matched calibration curve, which was obtained by leastsquares linear regression analysis of the peak area of the analytes versus the corresponding concentration in the matrix solution. The determination coefficient (r2) was higher than 0.990 and deviations of the individual points from the calibration curve were lower than 20%.
![]()
Analyte
Calibration type
Linear range
Regression equations
Determination coefficient
Slop ratio*
(μg.L-1)
(r2)
Moenomycin A
Matrix-matched
20-2000
y=9.44×103x-1.53×104
0.9990
1.14
Solvent calibration
20-2000
y=8.30×103x-1.15×104
0.9998
*Slop ratio of matrix-matched standard calibration curve to standard solution calibration curve.
Table 2: Calibration curves comparison for matrix effects evaluation.
Turness and precision were estimated through recovery, repeatability (intra-day) and within-laboratory reproducibility (interday) studies, which were assessed by spiking blank kidney tissue samples at three concentration levels (10, 50, and 500 μg.kg-1) in six replicates at each level for three consecutive days. All the values were calculated from matrix-matched calibration curve with the average recoveries ranged from 67.2 to 89.4%, as well as the repeatability and reproducibility in the range of 4.2-6.8% and 8.3-10.6% respectively (Table 3).
![]()
Analyte
Spiking levels
Intra-day (n=6)
Inter-day (n=18)
(μg.kg-1)
Recovery (%)
Repeatability (RSD, %)
Recovery (%)
Reproducibility (RSD, %)
Moenomycin A
10
67.2
6.8
71.9
8.3
50
72.9
5.3
89.4
5.9
500
82.7
4.2
85.0
10.6
Table 3: Trueness and precision of the proposed method in the spiked kidney samples.
The LOD and LOQ were calculated by analyzing blank kidney tissue samples spiked at the level of 1, 2, 10, 20, 50 and 100 μg.kg-1. The calculated values were 0.25 μg.kg-1 and 0.8μg.kg-1 respectively, which were enough to meet the MRL requirements set by “Positive List” in Japan [17].
Applications to real samples
To check suitability of the method and monitoring possible residue moenomycin A in poultry tissues. In local breeding factory (Shandong province, China), kidney tissue samples were collected from chickens that were subjecting to feed additives containing moenomycin. Thirteen samples out of twenty samples showed noncompliant results. The highest concentration was up to 74.5 μg.kg-1. To confirm the non-compliant result, enhanced production spectrum was acquired using MRM-IDA-EPI mode. As shown in (Figure 4a), transition of 789.9/575.9 was acquired in the survey scan at the time window of moenomycina A and a dependent scan was triggered to record product ions of 789.9 according to the scan scope set in EPI experiment. From (Figure 4b) several product ions were clearly observed in the EPI spectrum. Confirmation was conducted by EPI spectrum matching against EPI library built with standard solutions. In (Figure 4c), the EPI spectra from sample and moenomycin A standard were well matched by fitting main product ions in two spectra correspondingly. The matching purity was 98.8 (higher of the value indicates better of the matching), which was calculated from software of analyst (version 1.6.1, SCIEX). According to Decision 2002/657/EC [22]. One parent ion with two product ions can earn 4 IPs (identification points) to confirm banned drugs. In this instance, 789 with 729, 575, 279 and 466, which were obviously seen in the EPI spectra of sample and standard, can offer enough IPs to confirm the presence of moenomycin A. Findings from the tests gave a strong recommendation to monitor the moenomycin A in poultry tissues routinely.
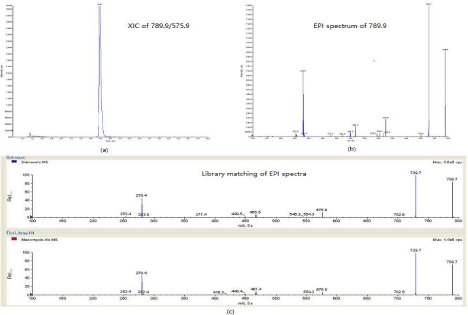
Figure 4: Confirmation analysis of Non-compliant sample with concentration of 74.5 μg.kg-1 in MRM-IDA-EPI mode: (a) Extracted ion chromatogram of 789.9/575.9
in survey scan; (b) Enhanced product ion spectrum of 789.9 triggered by survey scan; (c) EPI spectra matching against library built from standards.
Conclusion
As a feed additive has been used over 50 years, moenomycin shows its efficacy on weight gain and feed conversion rate in poultry farming. It also was considered as a safe antimicrobial feed additive with no residue concerns. But with banned regulations came into enforcement in EU and concerns due to prolonged feed exposure, overuse or inappropriate use of moenomycin, a selective and sensitive method is in demand for monitoring the possible remaining of moenomycin in poultry tissues, and if any, the performance of the method should meet requirements set by authorities, for instance, MRL specifications in “Positive List”. To our knowledge, it is the first time to report the detectable moenomycin A residue found in chicken kidney tissues using UHPLC-MS/MS method. Applications for real samples revealed that the proposed method could afford effective residue analysis of moenomycin A for routine and surveillance purposes.
Acknowledgement
The research was funded by the scientific and technological project of the General Administrative of Quality Supervision, Inspection and Quarantine of the People’s Republic of China (2013IK176), and was also partly supported by science and technology support plan of Qingdao (12-1-3-80-jh).
References
- Wallhausser KH, Nesemann G, Prave P, Steigler A. Moenomycin, a new antibiotic. I. Fermentation and isolation. Antimicrob Agents Chemother (Bethesda). 1965; 5: 734-736.
- Butaye P, Devriese LA, Haesebrouck F. Antimicrobial growth promoters used in animal feed: effects of less well known antibiotics on gram-positive bacteria. Clin Microbiol Rev. 2003; 16: 175-188.
- Welzel P. Syntheses around the transglycosylation step in peptidoglycan biosynthesis. Chem Rev. 2005; 105: 4610-4660.
- Halliday J, McKeveney D, Muldoon C, Rajaratnam P, Meutermans W. Targeting the forgotten transglycosylases. Biochem Pharmacol. 2006; 71: 957-967.
- Kurz M, Guba W, Vértesy L. Three-dimensional structure of moenomycin A--a potent inhibitor of penicillin-binding protein 1b. Eur J Biochem. 1998; 252: 500-507.
- Fehlhaber HW, Girg M, Seibert G, Hobert K, Welzel P, van Heijenoort Y, et al. Moenomycin A: a structural revision and new structure-activity reactions. Tetrahedron. 1990; 46: 1557-1568.
- Donnerstag A, Marzian S, Mueller D, Welzel P, Boettger D, Staerk A, et al. A structurally and biogenetically interesting moenomycin antibiotic. Tetrahedron. 1995; 51: 1931-1940.
- Scherkenbeck J, Hiltmann A, Hobert K, Bankova W, Siegels T, Kaiser M, et al. Structures of some moenomycin antibiotics-inhibitors of peptidoglycan biosynthesis. Tetrahedron. 1993; 49: 3091-3100.
- Hessler-Klintz M, Hobert K, Biallass A, Siegels T, Hiegemann M, Maulshagen A, et al. The First moenomycin antibiotic without the methyl-branched uronic acid constituent–unexpected structure activity relations. Tetrahedron. 1993; 49: 7667-7678.
- Ostash B, Walker S. Moenomycin family antibiotics: chemical synthesis, biosynthesis, and biological activity. Nat Prod Rep. 2010; 27: 1594-1617.
- Feighner SD, Dashkevicz MP. Subtherapeutic levels of antibiotics in poultry feeds and their effects on weight gain, feed efficiency, and bacterial cholyltaurine hydrolase activity. Appl Environ Microbiol. 1987; 53: 331-336.
- Mulder RW, van der Hulst-Van Arkel MC. Antibiotic residues in organs and muscle tissues of broilers. I. Bacitracin, flavomycin, spiramycin and viriniamycin residues following administration of diets containing low levels of these antibiotics. Tijdschr Diergeneeskd. 1976; 101: 1194-1198.
- Sambeth W, Nesemann G, Bauer F, Dost G. Investigations of the excretion and retention of flavomycin. In: Flovomycin Symposium. 1969; 133-139.
- Biro G, Lendvai I, Bokori J. Food hygienic aspects of poultry formula-feeds supplemented with antibiotics. Magyar Allatorvosok Lapja. 1983; 38: 95-98.
- Tseng HC, Chen LM, Yang CP. Residues of antibiotic feed additives in the tissues of broilers fed on the level of growth promotion and disease prevention. Journal of the Agriculture association of China. 1980; 112: 44-53.
- European Commission. Regulation 1831/2003/EC of the European Parliament and of the council on additives for use in animal nutrition. Off J Eur Commun. 2003; L268: 29.
- Ministry of Health, Labour and Welfare. The maximum residue limits of substance used as ingredient of agricultural in foods. MHLW Notification, No.370, 1959, amendment. 2005; 499.
- Salvatore MJ, Katz SE. Unified procedure for the determination of antibiotics in animal feeds. J AOAC Int. 1993; 76: 514-525.
- Gafner JL. Identification and semi quantitative estimation of antibiotics added to complete feeds, premixes, and concentrates. J AOAC Int. 1999; 82: 1-8.
- Gallo P, Fabbrocino S, Serpe L, Fiori M, Civitareale C, Stacchini P. Determination of the banned growth promoter moenomycin A in feed stuffs by liquid chromatography coupled to electrospray ion trap mass spectrometry. Rapid Commun Mass Spectrom. 2010; 24: 1017-1024.
- Pérez S, McJury BE, Eichhorn P, Aga DS. Determination of the antimicrobial growth promoter moenomycin-A in chicken litter. J Chromatogr A. 2007; 1175: 234-241.
- Commission Decision of 22 December 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRPLs) for certain residues in food of animal origin, Official Journal of European Union. 2004; L6: 38-39.
- Berendsen BJA, Stolker AAM, Nielen MWF. Selectivity in the sample preparation for the analysis of drug residues in products of animal origin. Trends Anal Chem. 2013; 43: 229-239.
- Lantzsch G, Binder H, Heerklotz H, Welzel P, Klose G. Aggregation behavior of the antibiotic moenomycin A in aqueous solution. Langmuir, 1998; 14: 4095-4104.
- Pfaller MA. Flavophospholipol use in animals: positive implications for antimicrobial resistance based on its microbiologic properties. Diagn Microbiol Infect Dis. 2006; 56: 115-121.
- Hoff RB, Rubensam G, Jank L, Barreto F, Peralba Mdo C, Pizzolato TM, et al. Analytical quality assurance in veterinary drug residue analysis methods: matrix effects determination and monitoring for sulfonamides analysis. Talanta. 2015; 443-450.