
Editorial
Austin J Clin Cardiolog. 2014;1(2): 1018.
Role for Mitochondria in ndothelial Function and Cardiovascular Diseases
Yukio Shimasaki*
Global Medical Affairs Japan Department, Takeda Pharmaceutical Company Limited, Tokyo, Japan
*Corresponding author: Yukio Shimasaki, Global Medical Affairs Japan Department, Takeda Development Center Japan, Takeda Pharmaceutical Company Limited, 12-10, Nihonbashi 2-chome, Chuo-ku, Tokyo 103-8668, Japan
Received: March 10, 2014; Accepted: March 20, 2014; Published: March 24, 2014
A Wide Variety of Functions and Distinguishing Features of Mitochondria
Mitochondria are evolutionary remnants of aerobic bacteria that invaded protoeukaryotic cells a billion years ago. The ability to utilize oxygen drove the development and evolution of the cardiovascular system in multicellular organisms, and for this reason mitochondria are linked to the cardiovascular system from the earliest stages of evolution [1]. Their role in the cardiovascular system is an intense area of recent research contributing to the idea that mitochondrial dysfunction is essential to cardiovascular disease (CVD).
Mitochondria are considered to be the powerhouse of the cell being the primary generators of adenosine triphosphate (ATP) (Figure 1). Known mitochondrial functions extend beyond ATP production to include other cellular processes such as mitochondriaderived reactive oxygen species (ROS) signaling, Ca2+ signaling, apoptosis, heme synthesis [2], inflammation [3], and embryonic development [4]. Interestingly, mitochondria communicate with the endoplasmic reticulum (ER) because the ER is the largest consumer of mitochondrial ATP and mitochondria critically depend on ERderived Ca2+ [5,6]. The interorganelle communication is importantfor the vital exchange of lipids, Ca2+, and ATP.
In addition, mitochondria communicate a great deal with the nucleus to enable mitochondrial biogenesis, which occurs in response to various stimuli, from cold to fuel deprivation to exercise. The peroxisome proliferator–activated receptor–γcoactivator–1α (PGC– 1α) is a master regulator of mitochondrial biogenesis. It has been argued that PGC–1α–induced biogenesis protects against excessive mitochondrial ROS and oxidative stress by supplying undamaged mitochondria that produce less ROS [7].
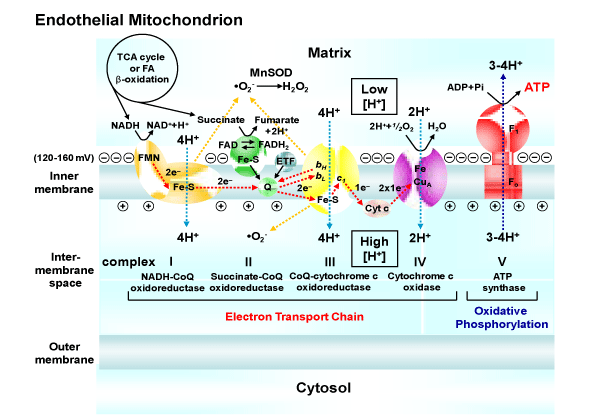
NAD: Nicotinamide Adenine Dinucleotide; FMN: Flavin Mononucleotide; Fe-S: Iron-Sulfer Cluster; FAD: Flavin Adenine Dinucleotide; ETF: Electron Transfer Flavoprotein; Q or CoQ: Coenzyme Q10; ‘bH, bL, c1: cytochrome bH, cytochrome bL, cytochrome c1; Cyt c: cytochrome c; ADP: Adenosine Diphosphate; Pi: Inorganic Phosphate; ATP: Adenosine Triphosphate; F1: Catalytic and Soluble Portion; Fo: Channel and Membrane Embedded Portion of ATP Synthase; �O2-: Superoxide; and MnSOD, Manganese Superoxide Dismutase.
Figure 1: The functional features of the mitochondrial respiratory chain and oxidative phosphorylation system. The electron transport chain carries both protons and electrons, passing electrons from donors to acceptors (e.g., NADH to O2), and transporting protons across a membrane.NAD: Nicotinamide Adenine Dinucleotide; FMN: Flavin Mononucleotide; Fe-S: Iron-Sulfer Cluster; FAD: Flavin Adenine Dinucleotide; ETF: Electron Transfer Flavoprotein; Q or CoQ: Coenzyme Q10; ‘bH, bL, c1: cytochrome bH, cytochrome bL, cytochrome c1; Cyt c: cytochrome c; ADP: Adenosine Diphosphate; Pi: Inorganic Phosphate; ATP: Adenosine Triphosphate; F1: Catalytic and Soluble Portion; Fo: Channel and Membrane Embedded Portion of ATP Synthase; "O2-: Superoxide; and MnSOD, Manganese Superoxide Dismutase.
Mitophagy refers to the selective autophagy (i.e., a regulated pathway of cellular degradation) of mitochondria. Stimuli for mitophagy also activate PGC–1α and biogenesis, providing fresh replacements for eliminated mitochondria [8]. Mitochondrial biogenesis, fission and fusion dynamics, and mitophagy provide a unified mechanism for mitochondrial quality control.
Thus, mitochondria have the capacity to impact a wide range of cellular processes via energy–dependent and energy–independent mechanisms.
Role of MitochondrialROS in Endothelial Cells
Mitochondrial content in endothelial cells is relatively modest, and energy requirements in these cells are relatively low compared to cardiac myocytes and other cell types. Endothelial cells obtain a large proportion of their energy from the anaerobic glycolytic metabolism of glucose [9,10]. Even if the endothelial mitochondrial respiration chain were to play a limited role in energy production, it may play a physiologically relevant role by virtue of its ROS–producing capacity.
Whereas the primary site of oxygen consumption in mitochondria is cytochrome c oxidase (complex IV), sources of mitochondrial ROS are complexes I and III. The first line of defense against mitochondrial superoxide is manganese superoxide dismutase (MnSOD), which catalyzes the conversion of superoxide anion to hydrogen peroxide. Levels of hydrogen peroxide are regulated by catalase and several mitochondrial and cytosolic peroxidases. Mitochondrial uncouplingproteins (UCPs) are considered another regulator of ROS production, and the importance of UCP2 in endothelial cells is confirmed by several experimental studies (11–13). For example, UCP2 impacts endothelial phenotype such as proliferative response via superoxide⁄ p53–mediated control of mitochondrial dynamics [13].
Nitric oxide (NO) is another important regulator of mitochondrial bioenergetics and oxidant production. Conditions that reduce endothelial NO bioavailability may promote excess mitochondrial ROS production by withdrawing the inhibitory effects of NO [14]. Under such conditions, peroxynitrite formed by the reaction of NO and superoxide may modify complexes I and III in a manner that further increases ROS production [15]. Antioxidant enzymes, including MnSOD, are also inactivated by peroxynitrite, further exacerbating oxidative stress within mitochondria [16].
Role for Mitochondria in Endothelial Function
Recent work has emphasized the importance of mitochondria in endothelial function as well as endothelial cell response to environmental cues. At any given time in endothelial cells, mitochondrial holds ˜25% of cellular calcium, although the ER is its major storage site [17]. Mitochondria serve as calcium buffer sites and regulate calcium uptake and release by the ER [5,18].
Cytosolic calcium levels regulate many aspects of endothelial function. For example, acetylcholine and serotonin activate endothelial NO synthase (eNOS) by increasing cytosolic calcium and stimulating the binding of calcium/calmodulin, leading to changes of eNOS gene expression and phosphorylation site, andactin cytoskeletal elements [19]. Calcium is also involved in vascular endothelial growth factor signaling [20].
Thus, mitochondria interact with the ER to regulate calcium signaling, whichis essential for the key aspects of endothelial function, including eNOS activation, motility, barrier function, and angiogenesis. A functional or structural disruption of the ERmitochondria unit could have multiple effects not only on calcium ignaling, but also on metabolism of lipids and ATP production.
Role for Endothelial Mitochondria in Diabetes and Cardiovascular Disease
The endothelium plays a central role in vascular homeostasis and the pathogenesis of CVD. Recent studies have shown that the severity of endothelial dysfunction relates to the risk for an initial or recurrent cardiovascular event. A growing number, but not all, of interventionsthat reverse endothelial dysfunction also reduce cardiovascular risk, and the ones known to reduce cardiovascular risk also improveendothelial function [21]. Endothelial dysfunction is thought to be a specific disease process of CVD.
Impaired calcium signaling between mitochondria and the ER causes eNOS dysfunction and NO bioavailability. Impaired NO signaling has been proposed as an important mediator of atherosclerosis [22]. In addition, endothelial mitochondria increase production of ROS in response to lipid oxidation products such as oxidized low–density lipoprotein (oxLDL) which infiltrates into the arterial endothelium [23].
In vitro studies have determined that hyperglycemia inhibits eNOS activity and expression in aortic endothelial cells by increasing mitochondrial superoxide production [24]. Hyperglycemia may also affect mitochondrial Ca2+dynamics as suggested by the finding that culturing endothelial cells under hyperglycemic conditions results in increase [Ca2+]m after histamine–mediated [Ca2+]c signaling, possibly as a consequence of altered mitochondrial morphology [25]. A recent study showed mitochondrial fragmentation and an increase in fission–1 (FIS1), a mitochondrial fission protein, in freshly isolated endothelial cells from patients with diabetes mellitus [26,27]. Genetic polymorphisms in the optic atrophy protein 1 and mitofusin–2 genes (OPA1 and MFN2; mitochondrial fusion genes) are associated with hypertension [28,29].
These studies suggest links between mitochondrial dynamics and cardiometabolic diseases, and raise the possibility that interventions directed toward restoring normal mitochondrial dynamics might have therapeutic benefit.
References
- Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013; 75: 95-126.
- Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006; 1763: 723-736.
- Seth RB, Sun L, Ea CK, Chen ZJ. Identiication and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005; 122: 669-682.
- Chen H, Detmer SA, Ewald AJ, Grifin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003; 160: 189-200.
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda). 2008; 23: 84-94.
- Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004; 2004: re1.
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, ission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008; 1777: 1092-1097.
- Mai S, Muster B, Bereiter-Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and inluence lifespan. Autophagy. 2012; 8: 47-62.
- Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. 2006; 103: 5379-5384.
- Culic O, Gruwel ML, Schrader J. Energy turnover of vascular endothelial cells. Am J Physiol. 1997; 273: C205-213.
- Duval C, Nègre-Salvayre A, Dogilo A, Salvayre R, Pénicaud L, Casteilla L. Increased reactive oxygen species production with antisense oligonucleotides directed against uncoupling protein 2 in murine endothelial cells. Biochem Cell Biol. 2002; 80: 757-764.
- Lee KU, Lee IK, Han J, Song DK, Kim YM, Song HS, et al. Effects of recombinant adenovirus-mediated uncoupling protein 2 overexpression on endothelial function and apoptosis. Circ Res. 2005; 96: 1200-1207.
- Shimasaki Y, Pan N, Messina LM, Li C, Chen K, Liu L, et al. Uncoupling protein 2 impacts endothelial phenotype via p53-mediated control of mitochondrial dynamics. Circ Res. 2013; 113: 891-901.Wang J, Alexanian A, Ying R, Kizhakekuttu TJ, Dharmashankar K, Vasquez- Vivar J, et al. Acute exposure to low glucose rapidly induces endothelial dysfunction and mitochondrial oxidative stress: role for AMP kinase. Arterioscler Thromb Vasc Biol. 2012; 32: 712-720.
- Wang J, Alexanian A, Ying R, Kizhakekuttu TJ, Dharmashankar K, Vasquez- Vivar J, et al. Acute exposure to low glucose rapidly induces endothelial dysfunction and mitochondrial oxidative stress: role for AMP kinase. Arterioscler Thromb Vasc Biol. 2012; 32: 712-720.
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008; 102: 488-496.
- MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998; 37: 1613-1622.
- Wood PG, Gillespie JI. Evidence for mitochondrial Ca(2+)-induced Ca2+ release in permeabilised endothelial cells. Biochem Biophys Res Commun. 1998; 246: 543-548.
- Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007; 100: 1128-1141.
- Sessa WC. Regulation of endothelial derived nitric oxide in health and disease. Mem Inst Oswaldo Cruz. 2005; 100 Suppl 1: 15-18.
- Andrikopoulos P, Baba A, Matsuda T, Djamgoz MB, Yaqoob MM, Eccles SA. Ca2+ inlux through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenicfunctions of human endothelial cells. J Biol Chem. 2011; 286: 37919-37931.
- Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003; 42: 1149-1160.
- Napoli C, Ignarro LJ. Nitric oxide and atherosclerosis. Nitric Oxide. 2001; 5: 88-97.
- Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Oxidized LDL induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. Am J Physiol Heart Circ Physiol.2005; 289: H852-861.
- Srinivasan S, Hatley ME, Bolick DT, Palmer LA, Edelstein D, Brownlee M, et al. Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia. 2004; 47: 1727-1734.
- Paltauf-Doburzynska J, Malli R, Graier WF. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J Cardiovasc Pharmacol. 2004; 44: 423-436.
- Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011; 124: 444-453.
- Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013; 112: 1171-1188.” The link is
- Wang Z, Liu Y, Liu J, Liu K, Wen J, Wen S, et al. HSG/Mfn2 gene polymorphism and essential hypertension: a case-control association study in Chinese. J Atheroscler Thromb. 2011; 18: 24-31.
- Jin HS, Sober S, Hong KW, Org E, Kim BY, Laan M, et al. Age-dependent association of the polymorphisms in the mitochondria-shaping gene, OPA1, with blood pressure and hypertension in Korean population. Am J Hypertens. 2011; 24: 1127-1135.