
Case Report
Austin J Clin Case Rep. 2014;1(3): 1012.
Progressive Glioblastoma Multiforme in a Patient with Sickle Cell Beta Thalassemia, Observations Following Combining Valproic Acid with Hydroxyurea
Haque S, Kim H, Cheruvu S and Olowokure OO*
Department of Internal Medicine and Hematology Oncology, University of Cincinnati, USA
*Corresponding author: Olowokure OO, Department of Internal Medicine and Hematology Oncology, University of Cincinnati, 234 Goodman Street, Cincinnati, OH 45219, USA
Received: May 30, 2014; Accepted: June 12, 2014; Published: June 14, 2014
Abstract
Increased Fetal Hemoglobin (HbF) appears to ameliorate the clinical severity of sickle cell disease. Studies have shown that multiple short chain fatty acids can induce gamma globin expression with increased HbF production. There are no randomized trials evaluating the combination of valproic acid a short chain fatty acid and Hydroxyurea (HU), both of which can independently increase fetal HbF production.
Here we report our observation in a sickle cell beta thalassemia patient with infrequent sickle cell pain episodes, who was diagnosed with Glioblastoma Multiforme (GBM), underwent treatment for this, with a drastic increase in the number and frequency of her sickle cell pain episodes that was subsequently placed on a drug regimen that included the combination of low dose HU and valproic acid.
Case Presentation
A 33 year old female with sickle cell beta thalassemia who had a baseline Fetal Hemoglobin (HbF) and hemoglobin A2 of 7.7% and 5.8% respectively by High Performance Liquid Chromatography (HPLC), was worked up for new onset seizures, headache and left sided weakness. She had a MRI which was suggestive of a right sided Glioblastoma Multiforme (GBM). Prior to her presentation she was known to have approximately one sickle cell pain episode every 3 years. She was started on phenytoin and dexamethasone. Her GBM was partially resected but with residual disease. She then received adjuvant radiation therapy. During the course of radiation, she was observed to have increased seizures, as well as increased frequency and intensity of sickle cell pain episodes, with frequent Emergency Department (ED) visits. She had to be hospitalized about twice a month for 3 consecutive months. She initially received multiple blood transfusions and partial exchange transfusions to reduce the percentage of sickle hemoglobin, but despite these measures and even with the sickle hemoglobin less than 30-40% by HPLC she continued to have sickle related pain, warranting multiple ED visits.
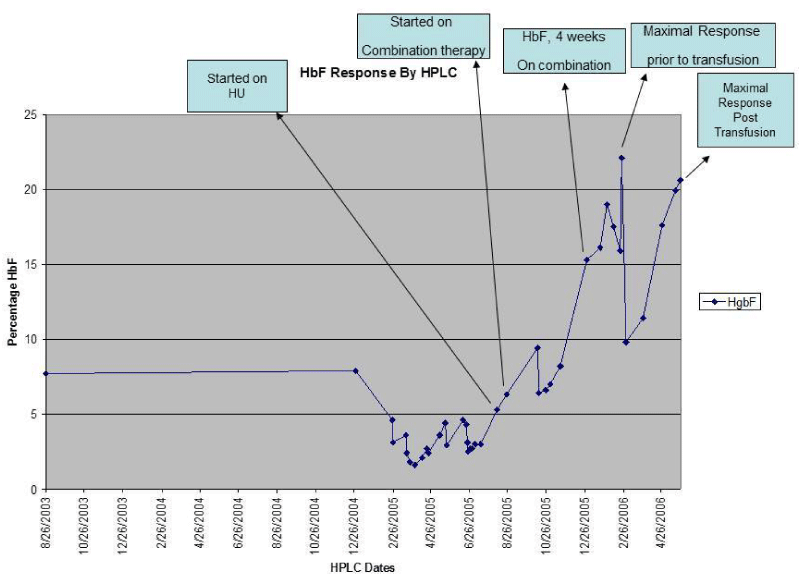
Figure 1 :Hbf by HPLC.
She was commenced on Hydroxyurea (HU) at a starting dose of 1 gram daily (14mg/kg/daily), which was reduced to 7mg/kg/day three weeks later, due to suspected toxicity. This was commenced in an attempt to raise fetal hemoglobin levels and decrease her need for transfusions. Two weeks after the initiation HU, patient was re-admitted to hospital for seizures, and valproic acid was commenced at a dose of 21mg/kg/day. After being started on valproic acid there appeared to be an improvement in sickle cell pain with no emergency room visits. She reported no significant pain at a 2 week follow up clinic visit, and continued to remain pain free 3 weeks later while on combined therapy, although she was also on pain medications. HPLC performed during her sickle cell clinic visits while on combination therapy for four weeks (Figure 1) and six weeks from the initiation of HU revealed an increase in HbF from 8.2% to 15.3% and a repeat measurement at her clinic visit one month later revealed an HbF of 19% which then increased to 22% three weeks later.
She was admitted with a pulmonary embolism and commenced on anticoagulation which was complicated by bleeding warranting a blood transfusion. Her post transfusion HbF declined to 9.8%. She continued on the same doses of her HU and valproic acid and was noted to have gradual increments of HbF on serial HPLC measurements, and by 12 weeks post transfusion her HbF was 20.6%.
She began to experience increased seizures with repeat head imaging showing continued tumor progression. She was not deemed a candidate for further surgical intervention and passed away 3 weeks after her last HPLC measurement.
Discussion
Increased Fetal Hemoglobin (HbF) appears to ameliorate the clinical severity of sickle cell disease [1]. The cooperative study of sickle cell disease a prospective study of the clinical course of sickle cell disease demonstrated that increased levels of HbF ameliorate some of the clinical manifestations of the disease and that levels greater than 9% could prevent early mortality from sickle cell anemia [2]. HU which suppresses bone marrow and increases HbF is the only FDA approved therapy to prevent sickle cell crises and was approved following results of the multicenter Clinical study of HU which demonstrated a significant reduction in the incidence of vaso-occlusive crises and acute chest syndrome in patients who received HU. In that study, the HbF levels increased in approximately half the adults from a mean of 5.1% to a mean of 8.6% [3].
Reports from previous literature suggest that butyrate a short chain fatty acid can induce embryonic erythroid cells to increase production of HbF in vitro by inducing the gamma globulin geneleading to increased gamma-globin synthesis [4]. Subsequent animal studies confirmed that butyrate inhibits fetal to adult Hb switch in sheep [5], and can stimulate HbF production in baboons [6,7]. Liakopoulou et al further demonstrated that along with butyrate, valproic acid another short chain fatty acid also increased Hb F by inducing the gamma globulin gene and postulated that other drugs with short chain fatty acids may share this property [8]. Xu et al found that both HU and butyrate selectively increase gamma G expression and may have a synergistic effect on HbF production [9]. Atweh et al reported on the use of butyrate to induce HbF in 3 out of 5 patients who did not respond to HU, that were subsequently treated with high doses of intravenous arginine butyrate infusions (2000 mg/kg/ day) with all 3 patients expressing increased HbF levels above 20% in response to butyrate [10]. A subsequent abstract by Sutten et al, described a patient with sickle cell disease who developed pulmonary hypertension, in whom the addition of butyrate to HU resulted in a peak HbF level of 45% and a marked amelioration of pulmonary hypertension [11]. Atweh and colleagues also reported on 3 patients who were enrolled on a combination therapy protocol consisting of HU for several months followed by HU and butyrate with all 3 patients demonstrating a marked increase in their HbF levels after butyrate was added to HU [12]. This case report further supports the possibility that combined therapy with a short chain fatty acid and HU may lead to sustained increases in HbF as we saw a rise in the percentage of HbF up to 19% within 4 weeks of combined therapy and even though the HbF decreased following transfusions, she appeared to continue to respond to this combination with gradual increment of HbF from 9.8% to 11.4% 4 weeks later. Eight weeks post transfusion HbF was 17.6% and then up to 20.6% twelve weeks post transfusion. She unfortunately passed away prior to further planned measurements.
In our experience with HU, doses as low as 7 mg/kg/day have not resulted in such dramatic responses within such a short period of time. We cannot however entirely rule out the possibility that the observed HbF response in our patient was due to HU alone. Valproic acid and HU in combination may lessen the tendency for vaso-occlusive crises in view of the fact that both agents can independently induce HbF production. Observations by other physicians involved in the care of sickle cell patients, that might have patients incidentally on this combination would be interesting.
References
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984; 63: 921-926.
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994; 330: 1639-1644.
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995; 332: 1317-1322.
- Ginder GD, Whitters MJ, Pohlman JK. Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc Natl Acad Sci U S A. 1984; 81: 3954-3958.
- Perrine SP, Rudolph A, Faller DV, Roman C, Cohen RA, Chen SJ, et al. Butyrate infusions in the ovine fetus delay the biologic clock for globin gene switching. Proc Natl Acad Sci U S A. 1988; 85: 8540-8542.
- Constantoulakis P, Papayannopoulou T, Stamatoyannopoulos G. alpha-Amino-N-butyric acid stimulates fetal hemoglobin in the adult. Blood. 1988; 72: 1961-1967.
- Constantoulakis P, Knitter G, Stamatoyannopoulos G. On the induction of fetal hemoglobin by butyrates: in vivo and in vitro studies with sodium butyrate and comparison of combination treatments with 5-AzaC and AraC. Blood. 1989; 74: 1963-1971.
- Liakopoulou E, Blau CA, Li Q, Josephson B, Wolf JA, Fournarakis B, et al. Stimulation of fetal hemoglobin production by short chain fatty acids. Blood. 1995; 86: 3227-3235.
- Xu J, Zimmer DB. Differential regulation of A gamma and G gamma fetal hemoglobin mRNA levels by hydroxyurea and butyrate. Exp Hematol. 1998; 26: 265-272.
- Atweh GF, Sutton M, Nassif I, Boosalis V, Dover GJ, Wallenstein S, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999; 93: 1790-1797.
- Sutton M, Weinberg RS, Padilla M, Perrine SP,Atweh GF. Progressive pulmonary hypertension and pulmonary insufficiency in sickle cell patients who respond to hydroxyurea [abstract]. Blood.1999; 94; 1845.
- Atweh GF, Fathallah H, Weinberg RS: Reactivation of fetal globin genes with butyrate and hydroxyurea: new clinical and mechanistic insights. American Society of Hematology Education Program Book. 2003; 19-25.